










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.1 Lisboa mar. 2017
CASE REPORT
Lupus nephritis in a patient without evidence of systemic lupus erythematosus
Sofia G Cerqueira1, Rui A Castro1, Rui M Abreu1, Susana Sampaio2, Pedro Rodrigues‑Pereira3, Ana T Nunes3, Teresa MPR Morgado1
1 Department of Nephrology, Centro Hospitalar Tras‑os‑Montes e Alto Douro, Vila Real, Portugal 2 Department of Nephrology, Hospital de São João, Porto, Portugal 3 Department of Pathology, Hospital de São João, Porto, Portugal
ABSTRACT
Lupus nephritis is a common complication of systemic lupus erythematosus (SLE), usually developing early in the course of the disease. Almost all patients with lupus nephritis present with positive ANA and anti‑dsDNA, Hypocomplementaemia and renal manifestations (haematuria, proteinuria and/or renal failure). However, in a small number of patients, the initial presentation is lupus nephritis with absent serology or clinical features of systemic lupus erythematosus.
Keywords: Antinuclear antibody negative‑lupus, full‑house nephropathy, lupus nephritis.
INTRODUCTION
According to the American College of Rheumatology and Systemic Lupus International Collaborating Clinics, the diagnosis criteria for systemic lupus erythematosus (SLE) comprise clinical (cutaneous lupus, oral or nasal ulcers, alopecia, arthritis, serositis, renal or neurologic involvement, haemolytic anaemia, leukopaenia and thrombocytopaenia) and immunological (ANA, anti‑dsDNA, anti‑Sm, antiphospholipid antibody, hypocomplementaemia, and positive Coombs) parameters. Four criteria are needed to make a diagnosis, with at least one of these clinical, and one immunological. Nevertheless, in some cases, diagnosis can be established with a renal biopsy showing lupus nephritis, together with ANA and anti‑dsDNA.
Lupus nephritis is a common and potentially lethal complication of SLE. Around 23 to 60% of patients with SLE develop kidney manifestations in the first three years of the disease. Early diagnosis and prompt treatment reduce morbidity and mortality. Typically, the kidney biopsy shows a glomerular full house immunofluorescence staining (IgA, IgG, IgM, C3, C4, and C1q with ≥1+ intensity). Some patients present this pattern without clinical or serologic criteria for SLE (lupus‑like glomerulonephritis).
CASE REPORT
A 43‑year‑old man with nephrotic syndrome was referred to our Nephrology Department. Patients relevant medical history was chronic alcoholic disease, active smoking and recently diagnosed arterial hypertension. His deceased father had had end‑stage kidney disease of unknown aetiology. A sister suffered from Cushings syndrome. The patient had no previously known kidney pathology. He had been admitted to the Intensive Care Unit in October 2015 with sepsis and Acute Respiratory Distress Syndrome associated with bilateral pneumonia. At this occasion, haematoproteinuria was detected. Echocardiogram was negative for infectious endocarditis. A gradual resolution of the respiratory insufficiency allowed transference to the ward at day 8 and he was discharged at day 16.
In February 2016, during a routine appointment, nephrotic syndrome was detected (proteinuria 13g/24h, serum albumin 2.3 mg/dL, total cholesterol 437mg/dL and triglycerides 216 mg/dL). Urinary sediment showed 5‑10 erythrocytes per high‑power field, without cylindruria. It was only then that he was referred to our Nephrology Department.
His serum creatinine was 0.96 mg/dL. ANAs (<1/160) and ANCAs (<1/20), Hbs Atg and HCV were negative.
No hypocomplementaemia was detected (C3 = 100 mg/ dL, C4 = 23mg/dL). Serum IgG was low (383 mg/dL), but both IgA and IgM were normal. Immunoelectrophoresis showed no alteration. Biomarkers for malignancy were negative and thyroid function was normal.
Ultrasound showed normal‑sized kidneys, with regular contours and normal differentiation. No alterations were found in the medical imaging study – echography and CT of central nervous system, thyroid, thorax, abdomen, and prostate, and video endoscopy of the gastrointestinal system.
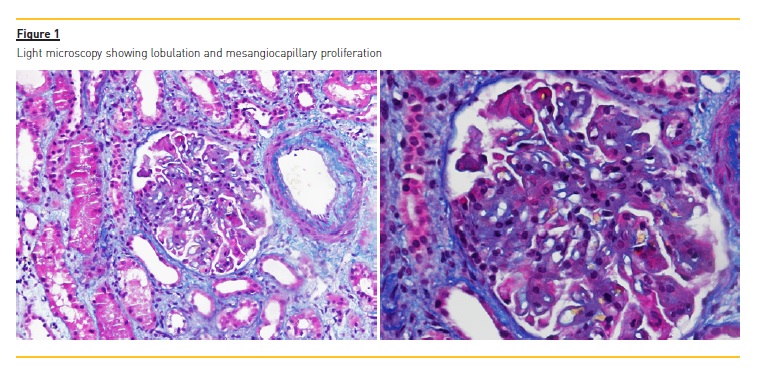
A kidney biopsy was performed in April 2016. It showed 17 glomeruli, 3 fully sclerotic, and another 3 with segmental sclerosis. In the light microscopy the glomeruli were lobulated, a mesangiocapillary proliferation as remarkable and a thickening of the glomerular basement membrane (no double contour), with sub endothelial and sub epithelial deposits was depicted (figure 1). Fibrinoid necrosis and crescents were absent.
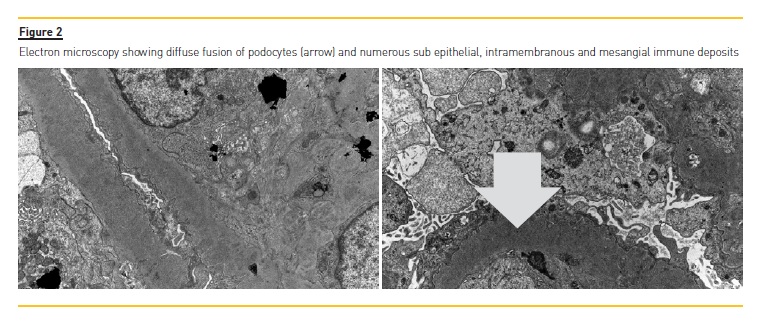
Tubular atrophy and interstitial fibrosis implicated around 25% of the sample. There was moderate inflammatory lymphocytic infiltrate of the tubules and interstitium, light hyaline arteriosclerosis and moderate atherosclerosis. Immunofluorescence showed a full house pattern (C1q, C3c, C4, IgG, IgA and IgM were all strong positive). Electron microscopy revealed diffuse podocyte fusion, thickening of the capillary basement membrane, expansion of the mesangial matrix, and also numerous sub epithelial, intramembranous and mesangial immune deposits (figure 2). Hence, a pattern of membranous and diffuse proliferative glomerulonephritis was described. Final histological diagnosis was lupus nephritis stage IV+V, index of activity 4, index of chronicity 4. Simultaneously, patients ANAs (<1/160) were negative and C3/C4 were normal (C3 97mg/dL and C4 21mg/dL).
Immunosuppressive therapy was initiated at that time, using oral mycophenolate mofetil 2g/day plus oral prednisone 1mg/kg/day. Unfortunately, on the 8th of August 2016 the patient developed a herpes zoster infection in his right arm, which was treated with oral acyclovir. At another routine Nephrology appointment, on the 17th of August, his laboratory data showed partial remission of proteinuria to 2.9 g/day, and sustained normal kidney function. Therefore, prednisone doses were reduced to 30 mg/day. On the 25th of August 2016, he was readmitted to the hospital ward due to worsening of the nephrotic syndrome and also a cutaneous bacterial superinfection of the herpes zoster area. At this time, immunosuppression was necessarily reduced.
At the last routine Nephrology appointment (October 2016), the proteinuria had climbed to 6.6 g/day but kidney function remained normal. Serology for SLE remained negative (ANA <1/160, C3 108 mg/dL and C4 29mg/dL). Then, mycophenolate mofetil was increased to 1g/day and prednisone to 0.5mg/kg/day. No other infections were noted thereafter.
DISCUSSION
Lupus nephritis is one of the major complications of Systemic Lupus Erythematosus. The pathognomonic immunological alterations arent always present at the time of diagnosis, challenging us in the differential diagnosis process.
Fortunately, the anatomical‑pathology markers of this disease are quite specific. For that reason, when present they should raise the suspicion of lupus nephritis, even in the absence of other serological or clinical criteria for SLE. The differential diagnosis comprises other entities with similar presentation, such as membranoprolipherative glomerulonephritis, post‑infectious glomerulonephritis, and imunotactoid/fibrillary glomerulonephritis.
The presence of typical signs of lupus nephritis at electron microscopy, namely tubuloreticular inclusions and also mesangial, sub endothelial and sub epithelial deposits, together with the typical findings on immunofluorescence are decisive to reach this diagnosis.
This disease should be distinguished from C1q nephropathy. In the latter, a pattern of glomerulonephritis with predominant mesangial C1q deposition on immunofluorescence and absence of tubuloreticular inclusions is present, but other histological features that resemble lupus nephritis may be present. However the typical SLE serology is negative and, also in contrast with lupus nephritis, C1q nephropathy is not responsive to immunosuppressive therapy. Sharmann et al. analyzed a series of 9 patients with presumed seronegative lupus nephritis. After 6 years of evolution showing no serological evidence of systemic lupus erythematosus and absent response to immunosuppressive therapy, the biopsies of these patients were reassessed, and afterwards their diagnoses was changed to C1q nephropathy.1
One should emphasize that, in our case, impressive proteinuria together with diminished serum IgG could impact on ANA titers into a false‑negative status.2
Seronegative lupus nephritis has an unspecific aetiopathogenesis and also a variable prognosis. Some cases can develop positive serology later in the course of the disease3‑6, after 6 to 10 years of follow‑up.
On the contrary, other subgroups of patients, mainly paediatric,7 maintained persistently negative SLE serology during 5 years of follow‑up.
Recently, a series of four adults with lupus nephritis was described. Their SLE serology was either persistently negative or presented low titer of ANA within 8 months to 4.5 years of follow‑up.8
Treatment and prognosis of lupus‑ like nephritis remain unclear, although limited data suggest a poor prognosis. Pirkle et al.10 presented the case of a 21‑year‑old female with lupus‑like nephritis, proteinuria of 1.2g/day and a positive lupus anticoagulant. She was treated conservatively with an aldosterone receptor antagonist class 2, and her proteinuria subsided, maintaining normal renal function after 4 months.
In a retrospective analysis by Ruggiero et al.11 of 42 children and adolescents with fullhouse nephropathy treated with immunosuppression, the great majority (76.2% of cases) did not show significant kidney failure or even develop complete remission of the renal dysfunction present at diagnosis, after a median of 5 years of follow‑up.
On the other hand, in the Huerta et al. series, with histology resembling lupus nephritis class IV, immunosuppressive therapy was ineffective. Yet, another identical case, described by Simmons et al.12, improved with this treatment.
Lupus‑like glomerulonephritis reports in the adult population are scarce. Moreover, there is limited experience in the follow‑up and treatment of this subset of patients. Most series published so far are mainly of paediatric age. Presently, there is no relevant information regarding the clinical presentation and evolution of this disease in adults. We present this case as an example of a rare disease that remains to be considered in the differential diagnosis of patients with this kind of presentation and requires specific and early treatment.
References
1. Sharmann A, Furness P, Feehally J. Distinguishing C1q nephropathy from lupus nephritis. Nephrol Dial Transplant 2004; 19: 1420‑1426 [ Links ]
2. Persellin RH, Takeuchi A. Antinuclear antibody‑negative systemic lupus erythematosus: Loss in body fluids. J Rheumatol 1980; 7: 547‑550 [ Links ]
3. Adu D, Williams DG, Taube D, et al. Late onset systemic lupus erythematosus and lupus‑like disease in patients with apparent idiopathic glomerulonephritis. Q J Med 1983; 52: 471‑487 [ Links ]
4. Cairns SA, Acheson EJ, Corbett CL, et al. The delayed appearance of an antinuclear factor and the diagnosis of systemic lupus erythematosus in glomerulonephritis, Postgrad Med J 1979; 55: 723‑727. [ Links ]
5. Baskin E, Agras PI, Menekşe N, Ozdemir H, Cengiz N. Full‑house nephropathy in a patient with negative serology for lupus, Rheumatol Int 2007; 27: 281‑284 [ Links ]
6. Nakahara, C et al, Delayed onset of systemic lupus erythematosus in a child with endothelial tubuloreticular inclusion, Clin Nephrol 2001; 56: 332‑335 [ Links ]
7. Gianviti A, Barsotti P, Barbera V, Faraggiana T, Rizzoni G. Delayed onset of systemic lupus erythematosus in patients with full‑house nephropathy, Pediatr Nephrol 1999; 13:340‑346 [ Links ]
8. Huerta A, Bomback AS, Liakopoulos V et al. Renal‑limited lupus‑like nephritis. Nephrol Dial Transplant 2012; 27: 2337‑2342 [ Links ]
9. Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol 2013; 24: 1357‑1366 [ Links ]
10. Pirkle JL, Freedman BI, Fogo AB. Immune complex disease with a lupus‑like pattern of deposition in an antinuclear antibody‑negative patient. Am J Kidney Dis 2013; 62: 159‑164 [ Links ]
11. Ruggiero B, Vivarelli M, Gianviti A, et al. Outcome of childhood‑onset full‑house nephropathy, Nephrol Dial Transplant 2016; gfw230. doi: 10.1093/ndt/gfw230 [ Links ] Simmons SC, Smith ML, Chang‑Miller A, Keddis MT. Antinuclear Antibody‑Negative Lupus Nephritis with Full House Nephropathy: a Case Report and Review of the Literature, Am J Nephrol 2015; 42: 451‑459 [ Links ] Fervenza SC, Sethi S. Evaluation and treatment of membranoproliferative glomerulonephritis, Literature review current through: Jul 2016. UptoDate. [ Links ]
Sofia Cerqueira
Department of Nephrology, Av. Noruega
Centro Hospitalar Tras‑os‑Montes e Alto Douro
5000 ‑508 Vila Real, Portugal
E-mail: ssacerqueira@gmail.com
Disclosure of potential conflicts of interest: none declared
All authors made substantial contributions to the conception and design of the manuscript, were involved in revising it critically for important intellectual content, and gave final approval of the version to be submitted
Received for publication: Nov 16, 2016
Accepted in revised form: Feb 21, 2017


{kind=link}
{kind=link}