










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.30 no.4 Lisboa dez. 2016
REVIEW ARTICLE
C3 glomerulopathies: A new category encompassing rare complement mediated glomerulonephritis
Fernanda Carvalho1, Fernando Nolasco1,2
1Laboratório de Morfologia Renal-Serviço de Nefrologia, Hospital Curry Cabral – CHLC
2 Nova Medical School - Faculdade de Ciências Médicas
INTRODUCTION
Disease associated membranoproliferative glomerulonephritis (MPGN) has been well -known for decades.
Recently, advances in the understanding of the complement system have shown its direct influence on MPGN-related conditions and conditions related to other morphologic entities. The key aspect is the clear demonstration of complement component C3 (C3) by immunofluorescence (IF) in glomeruli. A new group of rare diseases mediated by complement and called C3 glomerulopathies has emerged as part of renal conditions.
How to deal with a glomerulonephritis with predominant C3 deposits on immunofluorescence? The answer to this question is now much clearer as glomerulonephritis pathogenesis, histology and clinical aspects are better defined.
These complex entities are nowadays easier to understand, using a systematic approach and taking into account:
– the background knowledge of these diseases
– a better understanding of the complement role in this context
– a systematic way to diagnosis
– the choice of an appropriate treatment
– the perception of the evolution of these diseases after transplantation
BACKGROUND
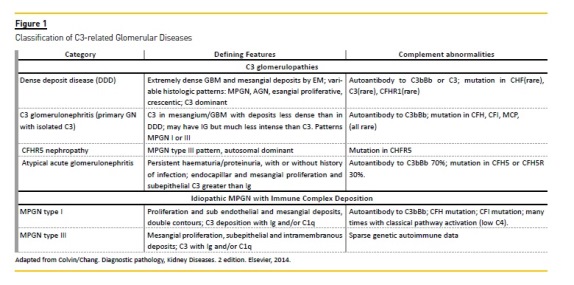
Going back to the 70s, MPGN classification was based on histological and electron microscopy (EM) findings1. Clinically they were classified as primary or secondary, depending on whether the aetiology was known or not. Primary or idiopathic diseases were subdivided into type I with mesangial and endocapillar proliferation accompanied by mesangial and subendothelial deposits creating double contours; type II where intramembranous dense deposits were the hallmark with variable proliferation and sometimes double contours, and type III similar to type I but with deposits in the subendothelial and subepithelial space.
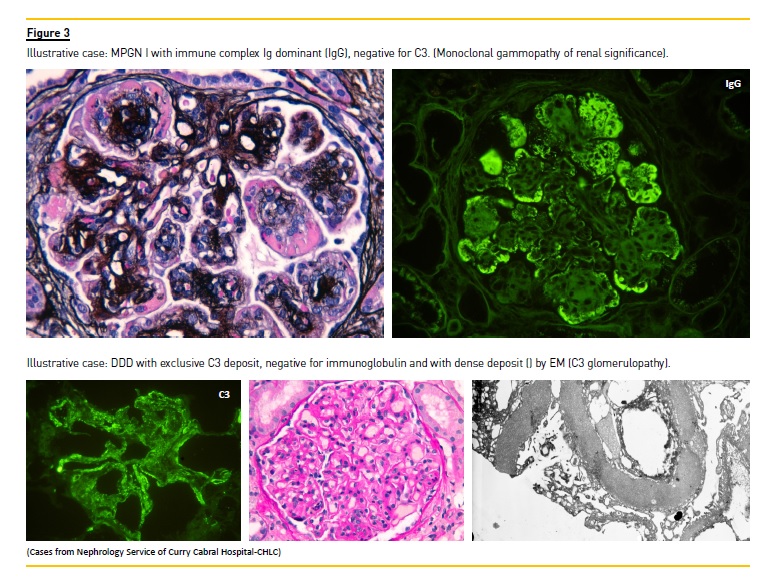
Renée Habibs group at Necker hospital, a group with an extensive experience in childrens biopsies worked to develop an improved immunofluorescence technique, achieving a very good differentiation of the patterns of immunoglobulin and C3 complement factor on glomeruli and tubular basement membranes of the biopsy specimen. C3 was dominant in type II MPGN with dense and linear deposits along the capillary walls and also with round deposits over the mesangium without the presence of immunoglobulin. EM revealed the dense deposit along the basement membrane and so the name of dense deposit disease (DDD) was introduced2.
DDD is part of the small and very rare group of diseases known today as C3 glomerulopathies.
The presence of immunoglobulin was the main fluorescence evidence in type I MPGN with little complement.
Low levels of serum complement were a well–known phenomenon although its role in the pathogenesis of these entities was largely unknown.
By 2011, immunofluorescence microscopy acquired a central role in the understanding of the MPGN pathogenesis.
A very special focus on composition and combination of the deposits in the glomeruli determined seen by IF3,4. MPGN type I shows deposits of immunoglobulin IgG/IgM/IgA and small amounts of complement C3. Diversely, MPGN DDD only shows deposits of C3, with small or no immunoglobulin deposits. Type III may show both deposits.
A more sophisticated interpretation of MPGN made by IF, by Vivette DAgaty et al. was published in 2014, comprising 319 cases of primary MPGN type I -III, representing a new look at MPGN5. The presence of immunoglobulin corresponds to type I/type III MPGN, and C3 alone is associated with MPGN type II. In this IF study the expression C3 dominant in the context of C3 glomerulopathy was proposed, as C3 is much more intense than any other deposit. These criteria imply investigation of the alternative complement pathway.
As in the past, pathologists diagnose dense MPGN deposits by EM and C3 IF deposits. Nevertheless, strong evidence showed that in an important number of cases the membranoproliferative aspects seen by light microscopy (LM) were absent, and definitely the designation of dense deposit disease - DDD - prevailed over MPGN type II.
While restricted C3 deposits were described by pathologists in MPGN type I and III, different morphological aspects by LM could be seen. The glomeruli could be almost normal, show mesangial or endocapillar glomerulonephritis and even show crescents. These GN with dominant C3 deposits and variable LM aspects are called C3 glomerulonephritis. This designation encompasses GN with different aspects by LM but with C3 and with no or small intensity immunoglobulin.
Prevailing C3 deposits in glomeruli are the basis of the electron-dense deposits image seen on EM. In some cases, these deposits show a very dense osmiophilic image which was given the name of DDD, whereas in other cases the deposits do not show this appearance and are designated C3 glomerulonephritis (C3GN). The distinction is not always clear-cut and an overlap of morphological and pathogenic aspects may occur.
COMPLEMENT
It is now established that complement regulation plays a central role in disorder pathogenesis such as MPGN and haemolytic uraemic syndrome (HUS). Its functional complexity is due to more than 30 proteins either circulating in plasma or in cell membranes.
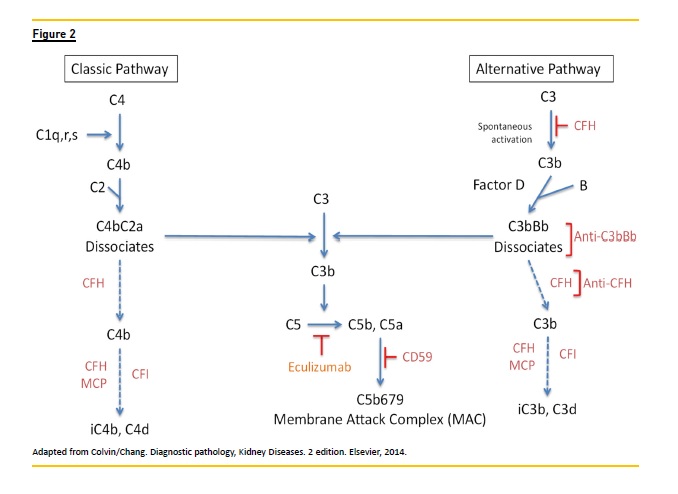
Complement activity depends of three pathways that rule our innate and acquired immunity. Those pathways act via C3 convertase on C3, giving origin to fragment C3a, an anaphylatoxin, and C3b, a potent opsonin. Rapid amplification of C3b can generate millions of C3b molecules within minutes. C3b with C3 originate C5 convertase which cleaves C5 in C5a and C5b. C5b triggers the terminal complement cascade, including C5b, C6, C7, C8, and C9 known as membrane attack complex (MAC). MAC a porelike, lipophilic protein complex, with C5b to C9 that inserts in cell membrane and results in cell lysis, Fig.2.
Presence of immunoglobulin and complement in a biopsy signals an immune -complex lesion, meaning that the classical pathway has been activated by an infectious, autoimmune, malignant associated disease as gammopathy. On the contrary, when the immunofluorescence pattern shows deposition of C3 alone, denotes that C3 has been activated in tissues by deregulation of the alternative pathway Fig.3. This pathway is constantly being produced in an slow manner (tickover), producing C3 and C3b that with complement factor B (CFB) produce fluid convertase C3bBb. Regulation of alternative pathway is permanently under the scope of multiple proteins in the fluid phase and tissues, including complement factors H (CFH) and I (CFI) and membrane cofactor protein (MCP).
Any alteration of these proteins breaks the control of this pathway transforming its low grade activity into a hyperactive phase.
This transformation is the base of C3 glome rulopathies. By 2012 these glomerulopathies had become individualized entities with a better known pathogenesis, morphology and clinical features. As in other circumstances, many doubts and differences persisted concerning the classification, diagnosis and how and when to treat them.
That same year a meeting, took place at Cambridge, UK where experts worked to obtain a consensus over these entities, on matters such as definition, systematization of complement investigations, and complement therapeutics6.
Most of the final conclusions were already known, but discussion and exchange of experiences from around the world, produced a better understanding and clarified the place of C3 glomerulopathies as part of glomerulonephritis.
Main conclusions of the 2012 consensus included definition, role of complement, therapeutics and behaviour after transplantation. Definition of C3 glomerulopathies was already consensual.
They constitute a new category of complement mediated diseases, with an unequivocal presence of glomerular C3 deposits, with little or no immunoglobulin or electron dense deposits present in glomeruli.
Another important remark is that this denomination encompasses a range of conditions independent of LM or EM findings.
Attention to complement activity is a focal point; C3 glomerulopathy and haemolytic uraemic syndrome (HUS) are diseases where the activation of an alternative pathway is the pathogenesis core of both entities. They share some alterations but they are different diseases.
In HUS complement activation occurs in vascular endothelium causing thrombotic microangiopathy and there are no glomerular deposits by EM or by IF. C3 is absent.
In C3 glomerulopathy, the activation of alternative pathway leads to C3 fragmentation with deposition in glomeruli without deposits of C1q or C4 or immunoglobulin.
In a few cases we can see small amounts of immunoglobulin. Morphology is further clarified and may present many different aspects by LM and by EM. Morphological features may depict mesangial proliferative GN, MPGN, proliferative endocapillar GN. All these may show crescents. Rarely glomeruli are normal by LM.
EM demonstrates deposits intensely osmiophilic and dense, along the glomerular basement membrane, identifying DDD. C3 glomerulopathies whose deposits do not have the same characteristics of DDD, namely density, are designated C3 glomerulonephritis.
Immunochemistry (IHC) or IF that allow demonstration of intense C3 deposits in the glomeruli of all C3 glomerulopathies are most outstanding in diagnosis.
When C5b -9 is present in the kidney, revealed by IHC/IF, this can be a marker of activation of alternative pathway and anti C5 therapy should be considered.
Nevertheless, presence of C5b-9 complex may occur in a normal kidney or many months after therapy, even when C5 is already normal in serum.
Proteins that make up the complement work in a coordinated way under strict control. Some alterations (acquired, hereditary or immunologic) of these proteins detected by serologic assays provide important information about the state of alternative pathway and give important diagnostic guidelines:
– Activation – reduced C3, normal C4, reduced factor B
– C3 turnover – C3 reduced, C3d augmented
– C5 turnover – C5 reduced, C5b -9 augmented
– Measurement of C3 nephritic factors
– Measurement of serum factor H - Is the major fluid-phase regulator of the alternative pathway
– Serum paraprotein detection – paraprotein may be the cause of uncontrolled activation
– Screening for CFHR5 – this mutation is a cause of C3 glomerulopathy.
Issues that must be considered in cases of special clinical and serological interpretation requiring a more refined interpretation are:
– Measurement of serum factor B – reduced (Factor B circulates in the blood; important in the formation of C3 convertase)
– Measurement of serum C5 – reduced in terminal pathway activation with some benefit from therapeutic C5 inhibition.
– Measurements of anti factor B autoantibody.
– Mutation screening of complement regulatory genes (CFH, CFB, CFI, and CD46 – membrane complement regulator that exist as multiple isoforms in a single cell type).
Recommendations for serological investigation, in all patients include:
– C3, C4, factor H levels, paraproteins,C3Neff
– Screening for CFHR5
– Other investigations case by case.
Conclusions of the consensus were published on 2013. C3 glomerulopathies including DDD and C3GN share some morphological, genetic and clinical features. The main differences between pathologic and genetic aspects were listed above.
Both entities also share clinical aspects. These are better known in DDD. C3 glomerulopathy without dense deposits, a recently discovered condition with distinct identity, represents a heterogeneous group in terms of pathogenesis and clinical course.
DDD has a poor prognosis. It is a disease of children and young adults. In a series from Nasr, 20087, 39% of the adults were over 60 years of age. Males and females are affected equally, although some studies show some female predominance. The clinical picture is similar to that in immune complex MPGN Type I. Almost all patients have proteinuria and haematuria. Nephrotic syndrome is present from 12% to 65% in different series from Nasr and Servais. Renal insufficiency is common at presentation and more common in adults. Hypertension may be seen at onset or in the course of the disease.
A minority of patients with DDD may present lipodystrophy, with low levels of serum C3 and positive C3NeF. Recently LU et al. described an association between DDD and type 1 diabetes mellitus that is not yet well clarified8.
For C3GN Servais9 states that mean age at diagnosis was 30 years, much higher than for DDD. 25% of patients were less than 16 years old. 27% of patients had nephrotic syndrome at presentation, compared with 38% with DDD and 65% with MPGN type I. Two thirds of the patients with C3GN had micro haematuria at the beginning of the disease. Significantly at presentation, 40% of C3GN had low C3 compared with 86% of low C3 in DDD.
There is a variable outcome according to the underlying aetiology. In the French series of Servais9 the mean time to ESRD was similar to MPGN and DDD with an overall 10 years survival of 63%.
80% of men progress to chronic renal failure and only 21% of women do. This difference is not yet justified or understood.
CFHR5 nephropathy is a familial form of C3 GN occurring in patients of Cypriot descent, with CFHR5 mutation10.
The major clinical feature is microscopic haematuria in 90% of young patients. Proteinuria appears later in older age and 75% that attain ESRF are male.
What can be done when a biopsy with dominant C3 in the glomeruli occur?
– First make sure that C3 is clearly dominant over the other immune reactants.
– Determine with certainty its localization: mesangial, over basement membrane or both, endocapillar, epimembranous.
– Integrate the IF aspects with the features of LM, EM, serological data and clinic.
– Elaborate a differential diagnosis between MPGN and post -infectious GN.
– EM must reveal the deposits. DDD shows dense deposits over the BM and in mesangium. C3GN presents less dense deposits in other localizations, mesangial, endothelial or epimembranous.
– If needed check for auto - antibodies and mutations.
THERAPEUTIC ASPECTS IN C3 GLOMERULOPATHY
C3 glomerulopathies have not an optimal treatment.
They are rare, there are no prospective randomized clinical trials and aetiopathogenesis was partially understood recently.
In the majority of patients DDD treatment is not evidence-based11.
Treatment includes corticosteroids, other immunosuppressant, antithrombolitics, anticoagulants and plasmapheresis.
Some series12 observed some benefits via a combination of immunosuppression and rennin-angiotensin blockage.
C3 in glomeruli may activate C5 and MAC C5b-9.
Inhibition of C5 level could ameliorate glomeruli injury in DDD. Eculizumab is a humanized antibody with high affinity to C5, blocking activation of C5a and C5b and subsequently the MAC complex.
In a study by Bomback et al13 six adults with C3 glomerulopathy were treated with Eculizumab.
After 12 months of therapy, 2 patients with DDD showed a significantly reduction in creatinine and another patient had reduced proteinuria.
One patient with C3GN showed reduction in creatinine and improvement in morphology.
MAC serum levels normalized and repeated biopsies showed less inflammation, according with a diminished synthesis of the chemotactic C5a.
The pattern of immunofluorescence for C3 and C5b-9 persisted.
Whom to treat. And what treatment? Does morphology predict a response to therapy? Do DDD and C3GN respond the same way?
Is therapy against complement unique? Are other anti-cellular immunosuppressive therapies of use against C3NeF and other antibodies? How long should the treatment be maintained?
Future therapy will point to other proteins in complement cascade, namely to C3 convertase inhibition, impairing C3 breakdown and its deposition in basement membranes and mesangium. Other proteins deserving attention are C5 or constituents of terminal complement.
TRANSPLANTATION
Recurrence of C3 glomerulopathy data is scarce. In patients with DDD recurrence is almost universal11.
C3GN recurrence in a recent study with 10 patients was 60%9. One case of CFHR5 recurrence has been described by Vernon. Recurrence occurred 46 days after transplantation14.
One C3GN case, of our own experience, recurred 3 months after transplantation (unpublished).
Servais paper signals that some patients with C3GN may develop thrombotic microangiopathy post transplantation7.
CONCLUSION
Nowadays C3 glomerulopathies are an individualized group of diseases with a better defined place in the spectrum of glomerulonephritis. Morphologic definition of C3 glomerulopathies is concise and based on an unequivocal demonstration of C3 in glomeruli. Diagnosis begins with that evidence and completes with the demonstration of dense deposits on EM, knowing that LM may be diverse. EM permits a sub-division in DDD and C3 GN according to deposit density and localization.
The dominant revelation of C3 in glomeruli implies an immediate effort to exclude other diseases with dominant C3 in glomeruli, namely post infectious glomerulonephritis.
Simultaneously a systematized study of the alternative complement cascade must be performed, checking the serum alterations of complement factors, the development of antibodies against complement proteins and the existence of mutations. Following this path we can reach a diagnosis and initiate an appropriate treatment, choosing among the options currently available, taking into account the best scientific literature.
Eculizumab is a potent anti C5 inhibitor, with an effective effect in certain cases of C3 glomerulopathies
Other therapies against fulcral proteins in the complement cascade, namely C3 convertase and terminal complement are being studied. Antiproliferative therapy with immunosuppression should be considered in cases bearing clear demonstration of antibodies against complement proteins.
Transplantation data are scarce but recurrence is high and the few reported cases seem to point to early recurrences.
Finally it must be recognized that our understanding of the role of the complement system in glomerular lesions are still in its infancy. For instance it is well known that C3b receptor (CR1, CD35) is present on the surface of normal podocytes, but its function and the importance of such presence remains to be defined.
However recent data suggests that it may protect podocytes from complement mediated injury15.
It is recognized that the access to complement assays requires referral to specialized laboratories. Taking this point into account we list laboratories offering some or all of the complement assays.
− In Lisboa: Serviço de patologia Clínica – Secção de Imunologia do Hospital de S.José – CHLC, tel. 218841875. All serologic measurements of complement factors including factors B, H, I.
− In Coimbra: Serviço de Hematologia Clínica, Unidade de Hemostase do Hospital Pediátrico de Coimbra – CHUC tel: 339488700 Extensão
– 13901. Antibodies against complement fact ors, mutations of complement factors and regulators.
References
1. Levy M, Gubler MC, Sich M et al. Immunopathology of membranoproliferative glomerulonephritis with subendothelial deposits (type I MPGN). Clin Immunol Immunopathol 1978; 10:477-492. [ Links ]
2. Habib R, Gubler MC, Loirat C et al. Dense deposit disease: a variant of membranoproliferative glomerulonephritis. Kidney Int 1975; 7:204-215. [ Links ]
3. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: pathogenic heterogrneity and proposal for a new classification. Semin Nephrol 2011; 31: 341-348. [ Links ]
4. Sethi S, Fervenza F. Membranoproliferative glomerulonephritis – a new look at an old entity. N Engl J Med 2012; 336:1119-1131. [ Links ]
5. Hou J, Markowitz G, Bomback A et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int 2014; 85:450-456. [ Links ]
6. Pickering M, DAgati V, Nester C et al. C3 glomerulopathy: consensus report. Kidney Int2013; 84:1079-1089. [ Links ]
7. Nasr SH, Valeri AM, Appel GB et al. Dense deposit disease: clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol 2009; 4:22-432. [ Links ]
8. Lu DF, Moon M, Lanning LD et al. Clinical features and outcomes of 98 children and adults with dense deposit disease. Pediatr Nephrol 2012; 27:773-781. [ Links ]
9. Servais A, Noël LH, Roumenina LT et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 2012; 82:454-464. [ Links ]
10. Gale DP, de Jorge EG, Cook HT et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 2010; 376:794-801. [ Links ]
11. Appel GB, Cook HT, Hageman G et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol 2005; 16:1392-1403. [ Links ]
12. Nasr SH, Valeri AM, Appel GB et al. Dense deposit disease: clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol 2009; 4:22-32. [ Links ]
13. Bomback AS, Smith RJ, Barile GR et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 2012; 7:748-756. [ Links ]
14. Vernon K, Gale DP, Jorge EG et al. Recurrence o complemente factor H -related protein 5 nephropathy in a renal transplant. American Journal of transplantation 2011; 11:152-155. [ Links ]
15. Java A, Liszewski MK, Hourcade DE et al. Role of complement receptor 1 (CR1; CD35) on epithelial cells: a model for understanding complement -mediated damage in the kidney. Mol Immunol 2015; 67:584-595. [ Links ]
Fernanda Carvalho
Laboratório de Morfologia Renal – Serviço de Nefrologia,
Hospital Curry Cabral – CHLC
Disclosure of potential conflicts of interest: none declared
Received for publication: Nov 11, 2016
Accepted in revised form: Dec 6, 2016


{kind=link}
{kind=link}