










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.30 no.3 Lisboa set. 2016
CASE REPORT
Chronic myelomonocytic leukaemia: a presentation with rare extramedullary involvement
Tiago Borges1, Inês Rêgo1, Jenny Badas1, Sofia Marques2, Hugo Ferreira2, Ricardo Pinto3, Jorge Oliveira1
1 Department of Internal Medicine
2 Department of Nephrology
3 Department of Clinical Haematology
Centro Hospitalar de São João, Porto, Portugal.
ABSTRACT
A 74‑year‑old male with a recent diagnosis of chronic myelomonocytic leukaemia (CMML) was admitted for rapidly progressive renal failure (RPRF), associated with gait impairment due to muscle weakness and pain in the lower limbs. After exclusion of pre‑renal and post‑renal causes of RPRF, the workup revealed monocytosis, high levels of inflammatory markers, hypergammaglobulinaemia, nephrotic proteinuria and high serum and urinary lysozyme levels. Renal biopsy confirmed the diagnosis of lysozyme‑induced kidney injury and CMML‑associated vasculitis. An electromyogram also revealed sensorimotor axonal polyneuropathy. The patient was started on prednisolone and azacitidine. Improvement of limb symptoms and a decrease in monocyte count, renal function values, inflammatory markers and proteinuria were subsequently seen. Although lysozyme levels are consistently elevated in CMML, lysozyme‑induced kidney injury is a rare cause of renal failure. Filtered lysozyme appears to act as a direct tubular toxin and lysozymuria has been proposed as a valuable tool for detection of tubular damage. Polyneuropathy secondary to CMML is also infrequent and may be due to autoimmune mechanisms. We describe a case of lysozyme‑induced kidney injury, vasculitis and axonal polyneuropathy, presumably secondary to CMML, in which prednisolone and azacitidine seem to have been helpful in treating extramedullary leukaemic involvement.
Key‑Words: Chronic myelomonocytic leukaemia; lysozyme; polyneuropathy; rapidly progressive renal failure; vasculitis.
INTRODUCTION
Chronic myelomonocytic leukaemia (CMML) is a hybrid disorder characterized by both myelodisplastic and myeloproliferative features. It mostly affects the elderly and about 75% of cases are diagnosed after 60 years of age3. Its diagnosis is based on the presence of peripheral monocytosis (> 1x109/L), absence of Philadelphia chromosome or BCR‑ABL fusion gene, absence of rearrangement of PDGFRA or PDGFRB, less than 20% blasts in the blood or bone marrow and dysplasia involving at least one myeloid lineage.
CMML usually presents insidiously with systemic symptoms such as fatigue, weight loss and fever, but extramedullary leukaemic involvement is rare. Skin, lymph nodes, spleen, prostate, pleura and pericardium are among the most commonly affected organs, while renal involvement has rarely been reported4.
Parenchymal infiltration of leukaemia cells, thrombotic microangiopathy, tumour lysis, chemotherapy toxicity and radiation injury are among the most common mechanisms that are responsible for leukaemia‑associated renal failure2. Specifically, CMML can also induce kidney injury by amyloid deposition, production of tumour necrosis factor‑α, increased production and excretion of lysozyme and direct infiltration of monocytes.
An association with vasculitis has also been described4. Paraneoplastic autoimmune phenomena are often encountered in these patients and should be distinguished from tissue infiltration by CMML, since a different therapeutic approach is warranted.
CASE REPORT
A 74‑year‑old male with CMML and no other known chronic disease was admitted to hospital because of rapidly progressive renal failure (RPRF). The patient had been well until approximately three months before admission, when he noticed ankle arthralgia with non‑pitting oedema but no other signs of arthritis. The symptoms persisted, along with loss of muscular strength in the lower limbs, asthenia, anorexia and generalized myalgia. About two months before admission, anaemia (haemoglobin 7.3 g/dL) was documented and, after a negative endoscopic screening, the patient was referred to an outpatient haematology clinic, where a blood smear was performed revealing granulocytic dysplasia and a monocytoid population (about 30%) with rare immature cells. A diagnosis of CMML type 2 was established and confirmed by bone marrow aspiration (21.9% monocytes, about 36% with markers of immaturity). Bone biopsy showed hypercellular marrow, high myeloid:erythroid ratio, left shift of myeloid maturation and numerous monocytoid cells, plasma cells and dysmorphic megakaryocytes, with an increased reticulin framework and abundant iron deposits; immunohistochemistry showed the presence of myeloperoxidase, lysozyme, CD34 and light chains, both κ and γ. The patient was started on supportive therapy with weekly blood transfusions. After the first transfusion, the patient mentioned an episode of haematuria, along with dyspnoea and productive cough with purulent sputum. One week before admission, he was started on cefixime, followed by resolution of respiratory symptoms, even though haematuria episodes recurred twice during the month before hospital admission and constitutional symptoms worsened. Blood chemistry performed in the outpatient clinic revealed worsening of renal function (creatinine at admission 4.9 mg/dL from a baseline value of 1.2 mg/dL) with no hyperkalaemia or acidosis, along with anaemia (haemoglobin 7.9 mg/dL), monocytosis (2.13x109/L) and elevation of acute‑phase reactants (C‑reactive protein 116.7 mg/L), so the patient was admitted to the internal medicine department.
At admission, his gait was severely impaired and he was restricted to a wheelchair; no oedema, high blood pressure or signs of arthritis were present. Pre‑renal and post‑renal causes of RPRF were first excluded.
Ultrasonography revealed normal kidney dimensions and high diffuse cortical hyperechogenecity, and excluded urinary tract dilatation; the urinary bladder showed no alterations. The patient was then started on intravenous fluid therapy. The laboratory workup at admission is shown in table 1. Noteworthy are not only the presence of hypergammaglobulinaemia with normal κ/γ ratio and no monoclonal component at immunofixation, but also high levels of inflammatory markers despite negative serial blood cultures. Furthermore, the autoimmunity profile (including complement levels and antinuclear, anti‑neutrophil cytoplasmic and anti‑glomerular basement membrane antibodies) was negative, while schizocytes were not detected in peripheral blood and no significant eosinophiluria was present; viral serological tests (HIV, HBV and HCV) were also negative despite cryoglobulin positivity at very low titres. On the other hand, serum and urinary lysozyme levels showed marked elevations. The transthoracic echocardiogram showed normal biventricular systolic function and no significant valvular disease or vegetations.
Urinary measurements revealed proteinuria in the nephrotic range (3.7g/24 h) with significant erythrocyturia (2988.6/μL).
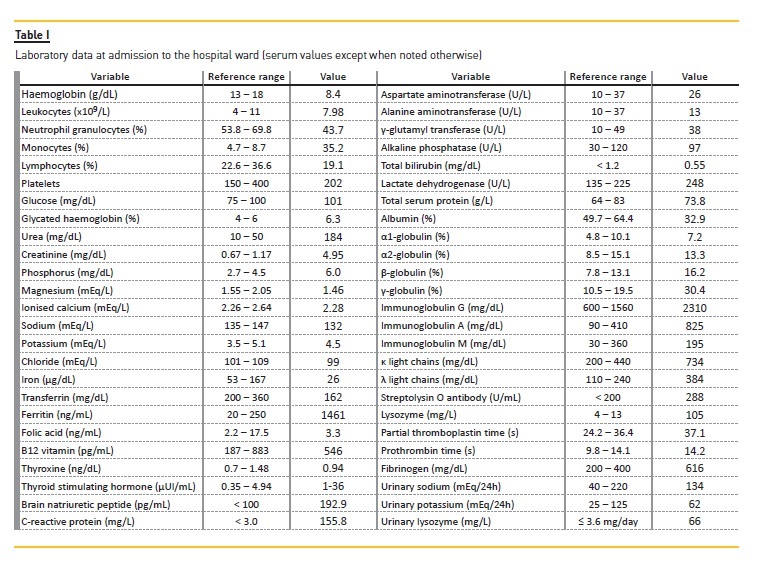
A slow improvement in renal function was observed during the first weeks (Fig. 1).
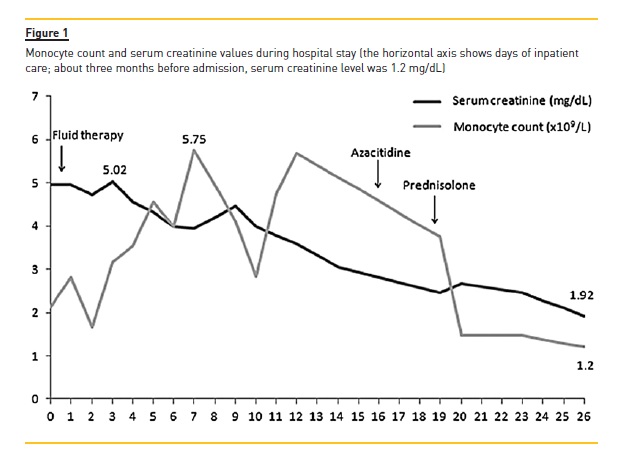
A renal biopsy was performed, revealing 23 glomeruli on optical microscopy: one with sclerosis, another with fibrinoid necrosis, three with cellular crescents, and another with mesangial proliferation and focal sclerosis. Immunohistochemistry analysis identified cells highly reactive for lysozyme in the capillary loops and peritubular capillaries, and variable reactivity in the luminal and tubular epithelial cells. Signs of tubular necrosis, haematuria and tubular atrophy were present, while fibrosis was observed in the interstitial space (about 25%), along with lymphoplasmocytic infiltrates but no evidence of leukaemic infiltration or amyloid deposition. A diagnosis of lysozyme‑induced kidney injury was established and electronic microscopy confirmed lysozyme deposits in the kidney (Fig. 2).
Electromyography was also performed, revealing sensorimotor axonal polyneuropathy; common causes of sensorimotor polyneuropathy, such as diabetes, alcoholism, vitamin B12 deficiency and autoimmune neuropathies were excluded (anti‑neuronal antibodies including anti‑Hu, anti‑Ri, anti‑Yo, anti‑amphiphysin and ganglioside antibodies were negative). The patient was started on prednisolone (20 mg/day) and azacitidine (75 mg/m2 subcutaneous).
Changes in serum creatinine values and monocyte counts during hospital stay are illustrated in Fig. 1; it should also be noted that after the first cycle of azacitidine, there were also decreases in proteinuria (0.88 g/24h) and C‑reactive protein (310.5 to 70.3 mg/L), along with improvement in limb symptoms, so that the patient was able to walk with assistance.
Two months after discharge, and when the prednisolone dose was gradually being tapered due to iatrogenic hyperglicaemia, the patient was readmitted to the hospital ward due to fever of unknown origin (FUO).
Azacitidine was stopped until diagnostic clarification, based on the clinical assumption that there was a high likelihood of uncontrolled infection and/or a high risk of CMML progression. The workup (including re‑test for anti‑neutrophil cytoplasmic antibodies and cryoglobulins, microbiological screening including bacterial, mycobacterial and fungal cultures of peripheral blood and bronchoalveolar lavage, along with cytological examination of bronchoalveolar lavage) was negative, except for a miliary pattern on thoracic computed tomography (CT) with multiple micronodular pulmonary lesions (Fig. 4). Despite therapy with broad‑spectrum antibiotics, antifungals and tuberculostatic agents, fever persisted and inflammatory markers remained high until high‑dose prednisolone (1 mg/kg/day) was restarted, when haemoglobin levels also increased and renal function values decreased. Thoracic CT was repeated one month later, and showed a significant reduction in the size of the pulmonary nodules (Fig. 3).
DISCUSSION
We describe a case of CMML in which renal impairment appears to be secondary to both vasculitic damage to the glomeruli and lysozyme‑induced tubular impairment.
In contrast to other types of leukaemia, combined glomerular‑tubular dysfunction appears to be characteristic of CMML12. Increased urinary and serum lysozyme may result in tubular dysfunction, but only rarely in glomerulopathy6. Lysozyme (or muramidase) is a bacteriolytic enzyme that catalyses the hydrolysis of the peptidoglycan layer of bacterial cell walls, and is found mainly in granulocytes, monocytes, macrophages and bone marrow precursors7. It was first discovered by Fleming in 1922, but it was only in 1966 that Osserman and Lawlor documented that CMML patients excreted large amounts of lysozyme in urine, making nephrotic‑range proteinuria relatively common in this setting8. In chronic myeloproliferative disorders, lysozyme levels are consistently high, while such elevations are usually restricted to monocytic forms in acute myeloid leukaemia7. Filtered lysozyme appears to be a direct tubular toxin, probably exacerbating ischaemic tubular damage2. Renal involvement in our patient was consistent with lysozyme‑induced kidney injury; however, in our case, not only was tubular damage present with lysozyme reactivity in tubular epithelial cells, but high reactivity was also observed in capillary loops and peritubular capillaries.
Measurement of urinary enzymes, such as lysozyme (lysozymuria), has been proposed as a valuable tool for detection of tubular damage, especially in the proximal tubules, and a good correlation between the degree of proteinuria and urinary lysozyme has been observed before treatment. Our patient presented high levels of both serum and urinary lysozyme. However, the value of lysozymuria in the clinical diagnosis of acute disorders of renal function is limited: increased serum lysozyme levels and the presence of lysozymuria may be indicators of severe kidney failure but are not sensitive markers in mild to moderate renal injury. Moreover, serum and urinary lysozyme may also be elevated in other diseases such as sarcoidosis, in which the former may be a more sensitive marker of disease activity than angiotensin‑converting enzyme2. Particularly in CMML patients, no correlation has been described between serum lysozyme levels and urinary levels of the enzyme or its renal clearance13. In haematological conditions like CMML, high plasma lysozyme levels are due to increased rates of synthesis, while in uraemic patients, serum lysozyme is elevated due to decreased rates of catabolism, since extra‑renal sites are capable of breaking down lysozyme only at a rate of about 15% compared to patients with intact renal function. Our patient presented both CMML and RPRF, and the presence of the latter may have been the main contributor to the high levels of serum lysozyme observed, as the monocyte count was lower than would be expected for CMML. Despite marked variability, correlations have been described between lysozyme levels and the number of circulating granulocytes and monocytes, while renal failure is expected to contribute to a significant increase in serum lysozyme levels13. In our case, a correlation between serum creatinine and monocyte count was not apparent as, during hospital stay, the observed decrease in serum creatinine appears to have been associated with the administration of fluid therapy, while the decrease in monocyte count was apparently related to the initiation of azacitidine and/or prednisolone (Fig. 3).
Glomerular involvement with crescentic glomerulonephritis has been described in myelodysplastic syndromes and CMML, possibly reflecting immune dysfunction.
In our patient, glomerular vasculitic involvement was suspected on renal biopsy due to the presence of cellular crescents and fibrinoid necrosis.
However, glomerulopathy was not the only apparent manifestation of autoimmunity in our patient, since the peripheral nervous system and lungs might have been involved.
The first case of CMML with polyneuropathy was described in 1989 by Maeda et al., associated with IgA‑paraprotein and κ light chain on immunoelectrophoresis and high serum and urine lysozyme levels.
As in our case, symptoms improved with prednisolone, so autoimmunity may have a role in its pathogenesis, even though we also used azacitidine as a treatment option17. Chronic inflammatory demyelinating polyneuropathy and systemic antineutrophil cytoplasmic antibody‑negative medium‑sized vessel vasculitis (polyarteritis nodosa‑type) have also been associated with CMML and a possible autoimmune pathogenesis. In a case series of systemic ANCA‑negative polyarteritis nodosa‑type vasculitis associated with CMML, most patients fulfilled the criteria for polymyalgia rheumatic and presentation was often with FUO19. Our patient met some of the 2012 European League Against Rheumatism/American College of Rheumatology criteria for polymyalgia rheumatica, including age ≥ 50 years, bilateral shoulder pain, abnormal CRP and/or ESR, morning stiffness and absence of rheumatoid factor or anti‑citrullinated protein antibody. On the other hand, in another case series, azacitidine seems to have been useful in autoimmune diseases associated with myelodysplastic syndromes, including vasculitis and neuropathy20.Nodular pulmonary lesions in patients with haematological malignancies can be caused by infectious or, less often, non‑infectious entities, including primary lung cancer, lymphoma, post‑transplantation lymphoproliferative disease, thromboembolism and leukaemic infiltrates21. Tuberculosis would be the most likely diagnosis when faced with a miliary pattern in an immunosuppressed patient like this one, but the absence of mycobacterial isolations and the persistence of fever despite antimycobacterial medication weaken the possibility of such a diagnosis. On the other hand, the resolution of the clinical picture after high‑dose steroids strengthens the possibility that in this patient vasculitis was not limited to the glomeruli. In a case series of systemic medium‑sized vessel vasculitis associated with CMML, diffuse bilateral interstitial pneumonitis has been described in a case presenting with FUO in which transpulmonary biopsy showed a perivascular infiltrate19.
The treatment for CMML patients is largely supportive but drugs like azacitidine and decitabine may have clinical activity, although their effect on kidney function is unknown2. Practice guidelines recommend that in patients with myelodysplastic‑type CMML with ≥ 10% blasts in bone marrow, azacitidine should be added to supportive therapy22. In our case, azacitidine and prednisolone appear to have had a positive effect not only on monocyte count and inflammatory markers, but also on renal function, proteinuria and neurological symptoms.
As kidney injury seems to have been mediated by both elevated lysozyme and vasculitis, we cannot assess the relative impact of each of these conditions on the patients lysozyme-induced kidney injury.
Finally, even though data are lacking on long‑term outcomes of lysozyme‑induced kidney injury, the prognosis of CMML is often worse than that for myelodysplastic syndromes in older adults and rates of transformation into acute myeloid leukaemia are variable (incidence of 15‑52%).
Factors that have been linked to worse outcomes in other studies, and that were present in our patient, include male sex, haemoglobin less than 10 g/dL and elevated lactate dehydrogenase1.
On the other hand, the presence of systemic vasculitis and/or cryoglobulins has been associated with a poorer prognosis in myelodysplastic syndromes24.
In conclusion, lysozyme‑induced kidney injury and CMML‑associated vasculitis are relatively rare conditions and other causes of renal failure must be considered first. This case highlights the fact that lysozyme‑induced kidney injury may be independent of monocyte counts in peripheral blood and bone marrow. To our knowledge, there are only seven case reports of CMML causing renal failure; direct involvement is rare, only two of these considered renal failure to be due to lysozyme‑induced kidney injury and none to both lysozyme‑induced kidney injury and vasculitis.
References
1. Parikh SA, Tefferi A. Chronic myelomonocytic leukemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012;87(6):610‑619. [ Links ]
2. Patel TV, Rennke HG, Sloan JM, DeAngelo DJ, Charytan DM. A forgotten cause of kidney injury in chronic myelomonocytic leukemia. Am J Kidney Dis 2009;54(1):159‑164. [ Links ]
3. Robinson GT, Sundaram KR, Dilly SA, Bevan DH, Andrews PA. Renal failure in a patient with chronic myelomonocytic leukaemia. Nephrol Dial Transplant 1997;12(7):1500‑1502. [ Links ]
4. Hyams E, Gupta R, Melamed J, Taneja SS, Shah O. Renal involvement by chronic myelomonocytic leukemia requiring nephroureterectomy. Rev Urol 2009;11(1):33‑37. [ Links ]
5. Kobayashi K, Yokote T, Tsuji M, Takubo T, Inoue T, Hanafusa T. Renal infiltration associated with chronic myelomonocytic leukaemia. Br J Haematol 2009;147(4):414. [ Links ]
6. Morschhauser F, Wattel E, Pagniez D, et al. Glomerular injury in chronic myelomonocytic leukemia. Leuk Lymphoma 1995;18(5):479‑483. [ Links ]
7. Åström M, Bodin L, Hörsten P, Wahlin A, Tidefelt U. Evidence for a bimodal relation between serum lysozyme and prognosis in 232 patients with acute myeloid leukaemia. Eur J Haematol 2003;70(1):26‑33. [ Links ]
8. Osserman EF, Lawlor DP. Serum and urinary lysozyme (muramidase) in monocytic and monomyelocytic leukemia. J Exp Med 1966;124(5):921‑952. [ Links ]
9. Chavan S, Hase N, Chavan P. Urinary enzymes in nephrotic syndrome. Indian J Clin Biochem 2005;20(2):126‑130. [ Links ]
10. Wilkinson SP, Hirst D, Day DW, Williams R. Spectrum of renal tubular damage in renal failure secondary to cirrhosis and fulminant hepatic failure. J Clin Pathol 1978;31(2):101‑107. [ Links ]
11. Harrison JF, Parker RW, De Silva KL. Lysozymuria and acute disorders of renal function. J Clin Pathol 1973;26(4):278‑284. [ Links ]
12. Niwa T, Ito T, Matsui E, Ohta H. Serum and urinary lysozyme activities in patients with renal diseases. Tohoku J Exp Med 1974;114(1):27‑33. [ Links ]
13. Pruzanski W, Platts ME. Serum and urinary proteins, lysozyme (muramidase), and renal dysfunction in mono‑and myelomonocytic leukemia. J Clin Invest 1970;49(9):1694‑1708. [ Links ]
14. Hansen NE, Karle H, Andersen V, Ølgaard K. Lysozyme turnover in man. J Clin Invest 1972;51(5):1146‑1155. [ Links ]
15. Komatsuda A, Miura I, Ohtani H, et al. Crescentic glomerulonephritis accompanied by myeloperoxidase‑antineutrophil cytoplasmic antibodies in a patient having myelodysplastic syndrome with trisomy 7. Am J Kidney Dis 1998;31(2):336‑340. [ Links ]
16. Bogdanović R, Kuzmanović M, Marković‑Lipkovski J, et al. Glomerular involvement in myelodysplastic syndromes. Pediatr Nephrol 2001;16(12):1053‑1057. [ Links ]
17. Maeda T, Ashie T, Kikuri K, Ishyama N, Takakura M, Ise T. Chronic myelomonocytic leukemia with polyneuropathy and IgA‑paraprotein. Jpn J Med 1989;28(6):709‑716. [ Links ]
18. Isoda A, Sakurai A, Ogawa Y, et al. Chronic inflammatory demyelinating polyneuropathy accompanied by chronic myelomonocytic leukemia: possible pathogenesis of autoimmunity in myelodysplastic syndrome. Int J Hematol 2009;90(2):239‑242. [ Links ]
19. Hamidou MA, Boumalassa A, Larroche C, El Kouri D, Blétry O, Groulleau JY. Systemic medium‑sized vessel vasculitis associated with chronic myelomonocytic leukemia. Semin Arthritis Rheum 2001;31(2):119‑126. [ Links ]
20. Al Ustwani O, Ford LA, Sait SJ, et al. Myelodysplastic syndromes and autoimmune diseases – Case series and review of literature. Leuk Res 2013;37(8):894‑899. [ Links ]
21. Wingard JR, Hiemenz JW, Jantz MA. How I manage pulmonary nodular lesions and nodular infiltrates in patients with hematologic malignancies or undergoing hematopoietic cell transplantation. Blood 2012;120(9):1791‑1800. [ Links ]
22. Onida F, Barosi G, Leone G, et al. Management recommendations for chronic myelomonocytic leukemia: consensus statements from the SIE, SIES, GITMO groups. Haematologica 2013;98(9):1344‑1352. [ Links ]
23. Zandberg DP, Huang TY, Ke X, et al. Treatment and outcomes for chronic myelomonocytic leukemia compared to myelodysplastic syndromes in older adults. Haematologica 2013;98(4):584‑590. [ Links ]
24. de Holland A, Beucher A, Henrion D, et al. Systemic and immune manifestations in myelodysplasia: a multicenter retrospective study. Arthritis Care Res (Hoboken) 2011;63(8):1188‑1194. [ Links ]
Tiago Borges
Department of Medicine, Alameda Professor Hêrnani Monteiro
4200‑319 Porto, Portugal
E‑mail: mtiagoborges@gmail.com
All authors made substantial contributions to the conception and design of the manuscript, were involved in revising it critically for important intellectual content, and gave final approval of the version to be submitted.
Disclosure of Potential Conflicts of Interest: None declared
Received for publication: Jan 9, 2016
Accepted in revised form: Feb 20, 2016


{kind=link}
{kind=link}
{kind=link}
{kind=link}