










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.29 no.3 Lisboa set. 2015
CASE REPORT
Antenatal Bartters syndrome
Síndrome de Bartter antenatal
Rita Coutinho, 1Ester Pereira, 1Luisa Martins, 1Raquel Henriques, 1Eulalia Afonso1
1Department of Neonatology A, Centro Hospitalar e Universitario de Coimbra. Coimbra, Portugal
ABSTRACT
Bartters syndrome is a rare renal tubular disorder with an autosomal recessive pattern of inheritanceand an estimated prevalence of 1 per million people. There are two distinct clinical presentations: antenatal Bartters syndrome and classical Bartters syndrome. Antenatal Bartters syndrome is considered the more severe form having its inception in utero. The typical features include early onset of foetal polyuria causing maternal polyhydramnios and preterm delivery, intrauterine growth restriction, postnatal polyuria and dehydration, nephrocalcinosis and osteopenia. We herein report a case of a newborn with antenatal Bartters syndrome, early suspected due to the familial and antenatal history. A 31 weeks gestation baby was born from consanguineous parents. Pregnancy was complicated with polyhydramnios at 24 weeks of gestation. The newborn developed polyuria, dehydration, electrolyte imbalances and metabolic alkalosis. Additionally, secondary hyperaldosteronism and hyperreninaemia with normal blood pressure were found. His older brother, also born preterm, had nephrocalcinosis and hypercalciuria attributed to prematurity and its complications. A diagnosis of Bartters syndrome type 2 was made by molecular study, which helped to clarify the clinical picture of his older brother. Antenatal Bartters syndrome manifestations can be neglected and undercovered by the diagnosis of prematurity. Hence, obstetricians and paediatricians should be aware of this rare disorder. A complete and careful clinical history is of the utmost importance to reach a definitive diagnosis.
Key-Words: Antenatal Bartters syndrome; electrolyte imbalance; polyhydramnios; polyuria; prematurity.
RESUMO
A Síndrome de Bartter é uma doença renal tubular rara, de transmissão hereditária autossómica recessiva, com uma prevalência estimada de 1 por milhão de pessoas. Existem duas formas distintas de apresentação clínica: síndrome de Bartter antenatal e síndrome de Bartter clássico. A síndrome de Bartter antenatal é considerada a forma mais grave, tendo início in utero. As caraterísticas típicas incluem o início precoce de poliúria fetal condicionando polidrâmnios materno e parto prematuro, restrição de crescimento intrauterino, poliúria e desidratação pós natais, nefrocalcinose e osteopenia. Apresentamos o caso de um recém-nascido com síndrome de Bartter antenatal suspeitado precocemente devido à história familiar e pré-natal. A gravidez foi complicada às 24 semanas por polidrâmnios. Nasceu um prematuro de 31 semanas, filho de pais consanguíneos. O recém-nascido desenvolveu poliúria, desidratação, distúrbios hidroeletrolíticos e alcalose metabólica. Adicionalmente apresentava hiperaldosteronismo hiperreninémico, com tensão arterial normal. O irmão mais velho, nascido prematuro, tinha nefrocalcinose e hipercalciúria que foram atribuídas à prematuridade e às suas complicações. Foi feito o diagnóstico molecular de síndrome de Bartter tipo 2, o que ajudou a clarificar o quadro clínico do irmão mais velho. As manifestações da síndrome de Bartter podem ser descuradas e encobertas pelo diagnóstico de prematuridade. Desta forma, obstetras e pediatras devem estar cientes desta doença rara. Uma completa e cuidadosa história clínica é fundamental para chegar ao diagnóstico definitivo.
Palavras-Chave: Distúrbios eletrolíticos; polidrâmnios; poliúria; prematuridade; síndrome de Bartter antenatal
INTRODUCTION
Bartters syndrome is a rare renal tubular disorder with an autosomal recessive pattern of inheritance and an estimated prevalence of 1 per million people1.
The primary pathogenic mechanism is a defective transepithelial chloride reabsorption at the thick ascending limb of loop of Henle. As a result, hypokalemia, metabolic alkalosis and hyperreninaemic hyperaldosteronism occurs with normal blood pressure1-3.
There are two distinct clinical presentations: antenatal Bartter syndrome (ABS) and classical Bartter syndrome3. The former, also termed hyperprostaglandin E syndrome, is the more severe form having its inception in utero. The typical features include early onset of fetal polyuria causing maternal polyhydramnios and preterm delivery, intrauterine growth restriction, postnatal polyuria, episodes of dehydration, nephrocalcinosis and osteopenia2,3.
Molecular studies allowed identification of different subtypes of the syndrome, depending on genes involved in defective synthesis of proteins responsible for transport of various ions across tubular cells. Genes reported so far are SLC12A1(15q15-21) encoding the sodium-potassium-chloride co-transporter protein NKCC2 (type1), KCNJ1(11q21-25) encoding renal outer medullary potassium channel ROMK (type2), CICNKB (1p36) which encodes the chloride channel Kb (type3), BSND (1p31) encoding the common accessory B-SUBMIT Barttin for CIC-Ka and CICKb chloride channel (type4) and CASR gene (3q13) encoding the calcium ion-sensitive receptor CaSR (type 5)4. Types 1 and 2 are denominated ABS, because of their early clinical presentation. Type 3, also known as classic Bartters syndrome is detected during childhood and presents with growth delay, polyuria, polydipsia and anorexia. Type 4 is associated with neurosensorial deafness. Patients with type 5 develop parathormone deficit, as well as the symptoms that are common to all types of the syndrome5.
We herein present a case of ABS whose diagnosis was early suspected due to familial and prenatal history.
CASE REPORTS
A premature newborn male was born at 31 weeks and 3 days of gestation. He was the second child of consanguineous parents within the third degree, but otherwise healthy.
Pregnancy was complicated by severe polyhydramnios at 24 weeks, which led to hospitalization and therapeutic amniodrainage at 28 weeks of gestation.
Biochemical analysis of the amniotic fluid was not performed. Prenatal ultrasounds and foetal echocardiography were unremarkable, as well as maternal serologies and glucose tolerance test. Foetal karyotype was 46, XY normal. Spontaneous delivery occurred at 31 weeks gestation, made through caesarean section because of breech presentation. There was no need for reanimation (Apgar scores 9/7/9) and birth weight was 1830 g (appropriate for gestational age). The newborn was admitted at the intensive care unit because of prematurity.
The newborn had a 5-year-old sibling, born at 29 weeks. That pregnancy was also complicated by polyhydramnios at 25 weeks gestation. He had several complications in the neonatal period (acute renal failure, sepsis, intestinal perforation and resection), with hyponatraemia and hypokalaemia, requiring transient supplements of sodium and potassium chloride, but after this period electrolyte imbalances were not found. Afterwards, he was transferred and followed in a secondary hospital.
Later on, short stature, failure to thrive, nephrocalcinosis and hypercalciuria were detected. However, there was no clear diagnosis for those findings.
The newborn`s physical examination was normal, without dimorphisms or anomalies of the external genitalia, and he had normal blood pressure. Marked polyuria since the 4th day of life (D4) was observed, with a maximum urine output of 8 ml/kg/h, and several electrolytic imbalances requiring correction.
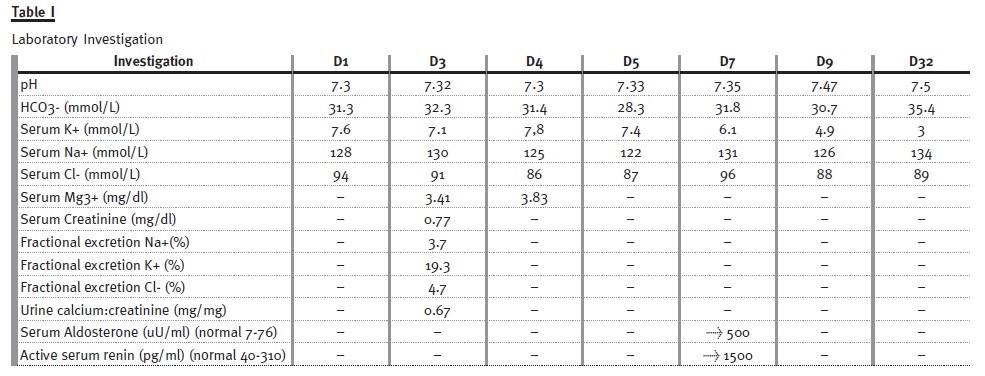
Maximum weight loss was 18% of the birth weight at D4 and birth weight was regained at D20. He presented hypochloremia, hyponatraemia, hyperkalemia up to D7, metabolic alkalosis after the first week of life and hypokalemia since D32 (Table I).
The fractional urinary excretion of sodium, potassium and chloride were increased and hyperreninaemic hyperaldosteronism was observed. The renal ultrasound at D3 was normal and at D36 was suggestive of macroscopic nephrocalcinosis (both renal sinuses had hyper-reflected spots); at 8 months medullary nephrocalcinosis was confirmed (the medullary pyramids were hyperechoic, bilaterally, and cortical thickness was preserved with no signs of hydronephrosis).
Molecular diagnosis for Bartters syndrome was performed, revealing the presence of a mutation c. 158T > G (p. Phe53Cys) in homozygosity in the KCNJ1 gene, described as pathogenic for Bartters syndrome type 2.
The treatment consisted in correction of dehydration and of electrolyte imbalances, with a maximum maintenance fluid of 200 ml/kg/day and of sodium 15 mEq/kg/day. Potassium supplements were introduced at D34. The baby was discharged at D38 with supplements of sodium chloride (10 mEq/Kg/day) and potassium chloride (2 mEq/Kg/day). On follow up, failure to thrive was observed, with an evolution for height and weight under percentile 3 based on the World Health Organization (WHO) child growth charts. Language delay was also detected, but with no changes regarding hearing acuity.
Currently, he is 21 months old and continues on a potassium supplement, having stopped the sodium supplementation at 4 months old. Until the present, indomethacin has not been introduced and the patient remains free of electrolyte imbalances.
DISCUSSION
The ABS is the severe form of Bartters syndrome with onset in utero3 and can be suspected and diagnosed before birth. Unexplained polyhydramnios between 24 and 30 weeks of gestation, without apparent foetal or placental abnormalities should lead to the suspicion of this entity3. Prenatal diagnosis can be made by documenting high chloride content of the amniotic fluid and mutational analysis of genomic DNA extracted from cultured amniocytes obtained by amniocentesis4,6. The mother can be treated antenatally with indomethacin, which improves the prognosis3,4. Indomethacin therapy and therapeutic amniocentesis usually allow the pregnancy to continue3. Unfortunately, in our case, despite having performed amniodrainage, biochemical analysis of the amniotic fluid, which would have led to an early diagnosis, was not made.
In our patient, the diagnosis of ABS was suspected early due to the family history and pregnancy antecedents: parents consanguinity; two pregnancies complicated by polyhydramnios and an older sibling born prematurely with nephrocalcinosis and hypercalciuria.
Postnatally, patients usually exhibit hyposthenuria and rapid weight loss. Major derangements are severe dehydration, hyponatremia, hypochloremia, hypokalaemia and metabolic alkalosis. Our case report presented all those features. On the other hand, we also observed transient hyperkalemia that may be also occur in type II ABS1,3,7. Despite the hyperkalaemia detected during the first week of life, it could have been confused with hypoaldosteronism or pseudohypoaldosteronism type I, the presence of a normal blood pressure was consentaneous with type II ABS.
Furthermore, nearly all ABS patients develop medullary nephrocalcinosis within the first month of life1, which was also observed by us.
Although presumptive diagnosis of ABS can be made on clinical and laboratory grounds, only the molecular study allows a definitive diagnosis and subsequent genetic counseling8. In our case, the molecular study confirmed the diagnosis of Bartters syndrome type 2 and helped to clarify the diagnosis of his elder brother, that albeit having the typical features of this syndrome in the prenatal and neonatal periods, the whole picture was attributed toprematurity and its complications, with impact in his management. Although the molecular study was not performed, he started indomethacin and potassium supplement, improving his appetite and weight.
There is no specific treatment, correction of dehydration and electrolyte imbalance are the important aspects of management. Generally, the use of prostaglandin synthetase inhibitors is required to control disease, indomethacin being the most recommended and well tolerated in children. Benefit from initiation of indomethacin therapy at 4-6 weeks with low doses (below 1 mg/kg/day) is more likely in patients with mutations at the ROMK channel gene7. In our patient, the early suspicion of diagnosis allowed the anticipation and more aggressive correction of dehydration and electrolyte imbalances, being aware that potassium supplements are usually needed by 2-3 weeks of age1,7,9. The newborn was discharged on the 6th week of life, so we decided not to start indomethacin because of the risk of necrotizing enterocolitis and acute renal failure before this age1,3,7.
The long-term prognosis is generally good, but close attention to electrolyte balance, volume status and growth is recommended. However, in a small minority of patients, hyperoxaluria, nephrocalcinosis and chronic indomethacin therapy can lead to chronic interstitial nephritis and chronic renal failure10.
This disorder is usually severe in the neonatal period and early childhood but it improves gradually which allows to reduce the intensity of treatment or stop it for a period1. The majority of children with a timely and appropriate therapy have clinical improvement and catch up growth. Long-term outcome including mental development is usually normal3.
CONCLUSION
The ABS is a rare entity but it should be suspected in any neonate with antenatal history of unexplained and rapidly increasing polyhydramnios in the late second trimester, that presents during the early neonatal period with polyuria and severe dehydration.
We wanted to leave a word of caution, namely to obstetricians and paediatricians as to the importance of making a complete and careful clinical history, because ABS manifestations can be neglected and mislead by the diagnosis of prematurity.
Early intervention and specific treatment options play a great role in reducing the complication rates of the affected neonates and should be initiated as soon as possible7.
References
1. Pérez JJL, Martinez LFJ, Alvarado EFG. Síndrome de Bartter. Reporte de un caso y revisión de la literatura. Rev Fac Med 2011;19(2):185-206. [ Links ]
2. Aloulou H, Ameur BS, Zouch I, et al. Neonatal Bartter syndrome type 1. Pediatric Oncall 2009;6(8):43. [ Links ]
3. Bhat YR, Vinayaka G, Sreelakshmi K. Antenatal Bartter syndrome: a review. Int J Pediatr 2012; doi: 10.1155/2012/857136. [ Links ]
4. Afzal M, Khan EA, Khan WA, et al. Antenatal Bartter syndrome. J Coll Physicians Surg Pak 2014;24 Suppl 2:S121-S123.
5. Higuita LMS, Londoño LMB, Vásquez CMM, Trujillo NP, Ruiz JJV. Síndromes de Bartter y Gitelman: revisión de los aspectos genéticos, fisiopatológicos y clínicos. Iatreia 2009; 22(1): 67-76. [ Links ]
6. Gupta M, Gupta S, Vasudev A, Srivastava RN. Neonatal Bartter Syndrome. Indian Journal of Clinical Practice. 2014 Oct; 25(5): 471-473. [ Links ]
7. Deniz NC, Agzikuru T, Akin Y et al. Neonatal Bartter syndrome: Case report. Early Human Development 2010, 86 (suppl):S53. [ Links ]
8. Reis GS, Miranda DM, Pereira PC, et al. Application of molecular biology at the approach of Bartters syndrome: case report. J Bras Nefrol 2012;34(1):82-86. [ Links ]
9. Amirlak I, Dawson KP. Bartter syndrome: an overview. QJM 2000; 93(4): 207-215. [ Links ]
10. Dell KM, Avner ED. Bartter-Gitelman syndromes and other inherited tubular transport abnormalities. in Nelson Textbook of Pediatrics. 18th ed. Kleigman RM, Behrman RE, Jenson HB, et al Eds Philadelphia: WB Saunders; 2007,2201-2202. [ Links ]
11. Devuyst O, Pirson Y. Genetics of hypercalciuric stone forming diseases. Kidney Int 2007;72(9):1065-1072. [ Links ]
12. Derst C, Konrad M, Köckerling A, et al. Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+ channel function. Biochem Biophys Res Commun 1997;230(3):641-645. [ Links ]
13. Finer G, Shalev H, Birk OS, et al. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr 2003;142(3):318-323. [ Links ]
14. Mendonça M, Pinheiro A, Castro E. Síndrome de Bartter: uma nova abordagem terapêutica. Acta Med Port 2011;24 Suppl 3: 671-674. [ Links ]
15. García Nieto V, Müller D, van der Vliet W, Claverie-Martín F. Enfermedad de Bartter neonatal diagnosticada mediante la detección de una mutación en el gen KCNJ1 que codifica la sínteses del canal renal de potássio ROMK. Nefrologia. 2001; 21(5): 448-54. [ Links ]
Drª Rita Coutinho
Maternidade Dr. Daniel de Matos
Rua Miguel Torga
3030-165 Coimbra, Portugal
E-mail: aritacoutinho@gmail.com
Conflict of interest statement: None declared
Received for publication: 08/06/2015 Accepted in revised form: 17/07/2015


{kind=link}