









Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.29 no.2 Lisboa June 2015
REVIEW ARTICLE
Regulation of erythropoietin production and recent trends in anaemia therapy
Regulação da produção da eritropoietina e perspectivas terapêuticas na anemia
Filipa Almeida, Sofia Santos, Idalina Beirão
Department of Nephrology, Centro Hospitalar do Porto. Porto, Portugal
ABSTRACT
About 30 years ago, the treatment of chronic renal disease anaemia was revolutionized by the introduction of recombinant human erythropoietin, which reduced the need for blood transfusions. In spite of this huge advance, the first recombinant human erythropoietin has a relatively short half-life and needs to be administered two to three times per week. Subsequently, other molecules were developed, such as darbepoetin alfa, continuous erythropoietin receptor activator (CERA) and peginesatide, with longer half-life, but the route of administration still remains a problem. Erythropoietin has an action that exceeds erythropoiesis and plays an important role in cell protection. Based on knowledge of the molecular mechanisms that control erythropoiesis, namely the regulation of EPO gene expression, through HIF system, GATA-2 and NF-kB, several upcoming therapeutic agents and strategies for stimulating and treating anaemia emerged.
The main effort in developing these treatments is to achieve other routes of administration, more convenient for the patient, such as oral therapy, not disregarding an easier production, storage and frequency of administration. Some of them are still in laboratory phase and others already in clinical trials phase II or III. In this work, based on a literature search of studies using MEDLINE, our objective is to review the regulation of erythropoietin production and its functions, as well as treatment approach for anaemia of chronic kidney disease, with particular focus on new therapies.
Key-Words: Anaemia; erythropoietin; GATA-2 inhibitors; hepcidin; hypoxia-inducible factors; kidney disease.
RESUMO
Há cerca de 30 anos atrás, o tratamento da anemia da doença renal crónica foi revolucionado pela introdução da eritropoietina (EPO) humana recombinante que permitiu reduzir drasticamente a necessidade de transfusões sanguíneas. Apesar deste grande avanço, a primeira EPO humana recombinante tem uma semivida relativamente curta e tem de ser administrada duas a três vezes por semana. Subsequentemente, foram desenvolvidas outras moléculas, como a darbepoetina alfa, o ativador contínuo do EPO-R (CERA) e o peginesatide, com uma semivida mais longa, mas a via de administração continua a ser exclusivamente parenteral. A eritropoietina desempenha várias funções além da eritropoiética. Tendo por base os mecanismos moleculares que controlam a eritropoiese, nomeadamente a regulação da expressão do gene da EPO, através do sistema do HIF, GATA-2 e NF-kB, surgiram vários fármacos e estratégias terapêuticas para o tratamento da anemia. O principal objetivo destes novos tratamentos passa por desenvolver outras vias de administração, mais cómodas para o doente, como a terapia oral, e facilitar a produção, armazenamento e frequência de administração dos fármacos. Alguns destes ainda se encontram em fase laboratorial, enquanto outros já estão em ensaios clínicos fase II ou III. Neste trabalho, baseado na revisão bibliográfica de artigos científicos publicados na MEDLINE, procuramos rever a regulação da produção da EPO e respetivas funções, bem como abordar o tratamento da anemia da doença renal crónica, com especial enfoque nas novas terapêuticas.
Palavras-Chave: Anemia; doença renal; eritropoietina; fatores induzíveis pela hipoxia; hepcidina; inibidores GATA-2.
INTRODUCTION
Erythropoietin (EPO), formerly named haemopoietin is essential to erythropoiesis. Since its discovery, much has been achieved in the knowledge of the biology of this glycoprotein. Currently, we know that EPO has an action that exceeds erythropoiesis and plays an important role in cell protection.
New erythropoietic agents designed to modulate the activation of the EPO gene are being studied.
Knowing that EPO has other functions, and also that regulators of the EPO gene are at the same time regulators of many other genes, we present a review on this issue. We will mainly focus on EPO production, regulation and functions, taking also into account current therapies and new options for anaemia treatment.
Erythropoietin
Erythropoietin is a hormone that controls erythrocytes production, promoting survival, differentiation and proliferation of erythroid progenitor cells in the bone marrow. Apoptosis prevention of these cells is the main mechanism underlying its function1. The human EPO gene, located on chromosome 7 codes for EPO, a protein composed of 165 amino acids, heavily glycosylated, with a molecular mass of about 30 kDa2. In adults, EPO is primarily synthesized in the kidney.
Previous studies showed conflicting results about renal EPO-producing cells (REPC), but the predominant location reported was the peritubular interstitial fibroblasts3 and tubular epithelial cells, mainly present in renal cortex (predominantly in the juxtamedullary region) and in outer medulla. Beirão et al. observed that distal nephron (epithelial distal tubular cells and collecting tubules) was the main locus of erythropoietin production in normoxic human kidneys4. In 2014, Nagai et al. reevaluated erythropoietin production and confirmed that erythropoietin is produced by cortical nephrons in normal haematopoietic condition, mainly by intercalated cells and not in peritubular cells as previously suggested. The production by peritubular cells occurred under hypoxic conditions, suggesting a different regulation mechanism between the nephrons and peritubular cells5.
During fetal life, EPO is produced by the liver. Similarly to the kidney, the liver responds to hypoxia increasing the number of EPO hepatocyte producers, located around the central vein. Erythropoietin was also found in liver stellate cells, previously called Ito cells6. In adult livers, EPO mRNA levels increase under moderate to severe hypoxia conditions, being one of the most important sources of extrarenal EPO, although insufficient to normalize haemoglobin in chronic kidney disease (CKD).
Apart from the kidney, EPO mRNA expression was also detected in non-haematopoietic tissues, such as brain (neurons and glial cells), lungs, heart, bone marrow, spleen, hair follicles, reproductive system, pancreatic islets and osteoblasts7,8. Under basal conditions, these cells do not play any role in erythropoiesis, but may contribute to induce stress erythropoiesis9.
In fact, EPO synthesized by these cells tends to act more locally, modulating, for example, regional angiogenesis and cell viability10.
Erythropoietin: Hematopoietic and cytoprotective functions
Erythropoietin is an endocrine, paracrine and autocrine hormone that acts as a cytoprotective hormone overextending its haematopoietic function. Among other effects, EPO antagonizes the activity of proinflammatory cytokines, has neuroprotective functions and promotes healing through stimulation of angiogenesis and capillary growth, has direct effects on immune, endothelial and bone marrow stromal cells, as well as on heart, brain, reproductive system, gastrointestinal tract, muscle, kidney, pancreas and nervous system cells7. Erythropoietin acts co-ordinately at various levels, which includes: limiting molecule production, such as reactive oxygen species and glutamate11, reversing vasospasm, attenuating apoptosis, modulating inflammation and recruiting stem cells12. It has other non-erythropoietic biological functions, like mitogenesis and angiogenesis, in part via endothelin-1 induction. Thus, anticipating an ischaemic insult (for example, kidney transplantation or renal arteries clamping during abdominal surgery), recombinant EPO can be used as a kidney tissue protector. Similarly, it may play an important role reducing CKD oxidative stress and vascular dysfunction.
Erythropoietin prevents tubular epithelial cells apoptosis and stimulates surviving cell population mitotic activity13. In the central nervous system, EPO is involved in neuroprotection, neurogenesis and angiogenesis, playing an important function as a neurotropic and immunomodulatory factor12. Angiogenesis promotes neurovascularization, allowing ischaemic zone revascularization and increased oxygen supply. At the same time, there is an increase in neural stem cells production and astrocytes and/or oligodendrocytes differentiation14. Erythropoietins protective effects were studied in vitro using adult rat cardiomyocytes and in vivo employing a rat model of coronary ischaemia–reperfusion. Erythropoietins anti-apoptotic activity reduces cardiomyocyte loss by about 50%, resulting in normalization of haemodynamic function. Due to cell death reduction, both compensatory hypertrophic response and inflammation are attenuated, preventing a misfit remodelling15.
In the skin, EPO enhances wound healing, reduces inflammatory response and increases capillary density in ischaemic regions. It promotes cardiac and central nervous system development, improves learning and memory, regulates angiogenesis, protects from ischaemia/reperfusion injury and retina from degeneration16.
It also protects against diabetes in mouse models, mediated by Janus Kinase-2 (JAK2) signalling directly in pancreatic cells, resulting in cell survival and proliferation, reduced inflammation and increased angiogenesis in the islets17. Erythropoietin may act on the regulation of metabolism and obesity and has potential benefits in the treatment of neurologic diseases, mood symptoms and depression (Fig. 1).
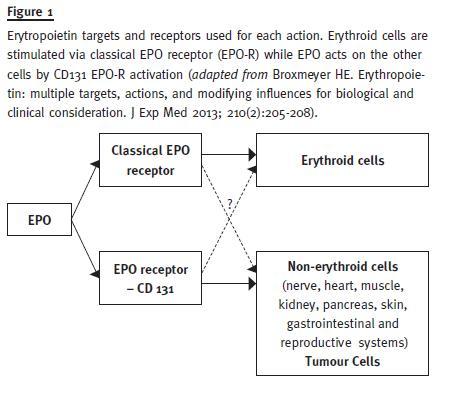
Therefore, EPO has an action that exceeds erythropoiesis and plays an important position in cell protection, using for that different cell receptors in erythropoiesis and cellular protection. This knowledge allowed the development of modified recombinant human EPO that only acts in cell protection, such as asialo-EPO, carbamylated EPO (CEPO) and neuro-EPO, a subject that goes beyond the scope of this work.
EPO gene regulation
Erythropoietin production is primarily stimulated by hypoxia and controlled transcriptionally. According to hypoxia severity, serum levels can increase up to several hundred times.
Hypoxia inducible factor (HIF) system: Once EPO binds to the EPO receptor (EPO-R) dimer, it induces JAK2 tyrosine kinase stimulation, leading to various proteins phosphorylation, including EPO-R itself.
Thus, different intracellular pathways are activated: signal transduction and activator of transcription 5 (STAT 5), phosphatidylinositol-3-kinase/Protein kinase B (PI-3K/Akt), Mitogen-Associated Protein Kinase/extracellular signal-related kinase (MAPK/ERK) and protein kinase C18. JAK2 activation also originates multiple binding sites for intracellular signalling proteins with src homology 2 (SH2) domains1. Erythropoietin enhancer (3) is activated by HIF, a heterodimeric protein that contains the basic helix-loop-helix domain and belongs to the transcription factors PAS [PER/aryl hydrocarbon receptor nuclear translocator (ARNT)/single minded (SIM)] family. It is composed of an O2-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit. Three α subunits are known: HIF-1α, HIF-2α and HIF-3α. Together with HIF-2α, HIF-1α facilitates oxygen delivery and cellular adaptation to hypoxia, stimulating a broad spectrum of biological processes. In fact, the number of known genes activated by HIF continues to increase and includes genes whose proteins are involved in angiogenesis, energy metabolism, erythropoiesis, cell proliferation and viability, mitochondrial biogenesis, vascular remodelling and vasomotor responses19.
Erythropoiesis and angiogenesis represent adaptive responses to improve tissue oxygenation, which require several days to develop – for example, the time required to the appearance of mature red blood cells in the circulation, mediated by EPO, is about a week. The HIF also mediates adaptive responses to hypoxia in a short term, regulating transcriptionally a large number of glucose transporters (GLUT-1,3) and glycolytic enzymes, as well as cell growth and survival genes (insulin-like growth factor 2 (IGF-2), insulin-like growth factor-binding protein (IGFBP) and transforming growth factor-α (TGF-α)20. Glycolytic enzymes induction demonstrates the role of HIF in the autonomous cellular adaptation to hypoxia, producing ATP by glycolysis, instead of oxidative phosphorylation.
The HIF activated genes are also involved in crucial aspects of cancer biology, such as angiogenesis, cell survival, glucose metabolism and invasion21 and HIF regulated genes are induced when HIF heterodimers bind to specific DNA sequences and transcriptional cofactors are recruited. These sequences are found in regulatory regions of various genes, oxygen sensitive, and are called the hypoxia response elements (HRE)2. In normoxic conditions, HIF subunits are rapidly degraded by proteasomes, after binding with von Hippel-Lindau tumour suppressor (VHL), which is the recognition substrate of the ubiquitin E3 ligase complex that mediates the ubiquitination of HIF. This ubiquitination prevents transcriptionally active heterodimer formation and requires hydroxylation of specific proline residues (Pro 402 and Pro 564 in HIF-1α; Pro 405 and Pro 531 in HIF-2α), which are located in oxygen-dependent degradation domains (ODD: lying on HIFα terminal C)22. The HIF inactivation by hydroxylation is carried out by three main 2-oxoglutarate (2OG) dependent oxygenases – prolyl-4-hydroxylase domain proteins (PHD) – PHD1, PhD2 and PHD3, which work as primary oxygen sensors, in the control of EPO production. The PHD are expressed in the kidney, where they control HIF activity23. In hypoxic conditions, hydroxylation is inhibited and HIF signalling is activated. A second HIF transcription pathway is related with factor inhibiting HIF (FIH). It is a 2OG oxygenase which catalyzes the hydroxylation of an asparginine residue, belonging to the transactivation domain of HIFα carboxyl terminal, that inhibits coactivators CREB-binding protein (CBP)/p300 binding to its transcriptional complex. Therefore, the opposite – HIF inactivation – facilitates CBP/p300 selection, resulting in increased HIF target gene expression, under hypoxia. In the kidney, FIH was detected in REPC, podocytes and distal tubule24. Although in vitro studies have identified HIF-1 as the responsible transcription factor for EPO induction in hypoxia conditions, HIF-2 has now emerged as the main regulator of EPO production, in vivo25. In fact, it is responsible for the prevention of erythroid progenitor cells apoptosis and the maintenance of a normal erythropoiesis, increasing erythrocytes production, under hypoxia10. Its transactivation in EPO HRE involves multiple nuclear factors that are associated with EPO gene. One of these factors is hepatocyte nuclear factor-4 (HNF4) that binds to the EPO enhancer, allowing interaction with HIF-2. As HIF-2, the cellular location of HNF4 expression corresponds with EPO production places in the kidney and liver. Low intracellular iron levels decrease HIF-2α translation, limiting both EPO production and erythropoiesis, when cellular iron stores are depleted, because, physiologically, erythropoiesis does not occur in the absence of iron. Besides HIF, EPO expression is also regulated by GATA-2 and nuclear factor kappa beta (NF-kB) at the level of the promoter. The EPO promoter (5) is suppressed by GATA-2 in normoxia conditions and, under hypoxia, GATA-2 levels decrease. The pro-inflammatory cytokines interleucine-1 (IL-1) and tumour necrosis factor-α (TNF-α) activate GATA-2 and NF-kB and may contribute to the anaemia of chronic disease in part by suppressing EPO production26.
Hepcidin: The EPO production in the kidney and liver stimulates erythropoiesis and an additional need for iron that leads to an increase in intestinal iron absorption and iron binding capacity, as well as an increase in iron release from body iron stores. The EPO production is coordinated with iron metabolism by hepcidin, the systemic iron homeostasis regulator, encoded by the HAMP gene. It is a small peptide consisting of 25 amino acids, mainly produced by hepatocytes, whose transcription is sensitive to iron and oxygen. In addition to its antimicrobial properties, hepcidin controls the amount of iron absorbed in the duodenum and the release of iron from the reticuloendothelial system cells (such as Kupffer cells and splenic macrophages) by ferroportin internalization and degradation, which is expressed in duodenal enterocytes, hepatocytes and macrophages27. The hepcidin regulation is complex, but one of the major stimulus for its production is IL-6 (via JAK/STAT transcription), produced as part of the inflammatory response, together with molecules like hemojuvelin and bone morphogenetic protein 6 (BMP-6). The hemojuvelin binds competitively to the BMP, which prevents the signalling of its receptor and suppresses hepcidin production28. In case of iron deficiency (such as iron deficiency anaemia) and/or hypoxic conditions, the liver decreases hepcidin production and intestinal iron absorption is enhanced. Increased serum hepcidin levels were observed in chronic situations frequently associated with inflammatory conditions, which reduces ferroportin expression and causes hypoferremia, supporting the key role of hepcidin in the pathogenesis of chronic disease anaemia29.
ANAEMIA OF CKD
Anaemia is defined as a haemoglobin concentration below 12 g/dl in women and less than 13 g/dl in men30. A large proportion of CKD patients develop anaemia over the course of their disease, which is a risk factor associated with worse prognosis, either as an independent predictor factor or as a risk multiplier in patients with concomitant cardiovascular disease31. Anaemia prevalence increases as renal function worsens to glomerular filtration rate lower than 60ml/min/1.73 m2 and is almost universal in stage 5 CKD32. Anaemia severity is related to glomerular filtration loss degree, but independent of kidney disease aetiology.
The CKD anaemia is hypoproliferative and normocytic normochromic, unless an iron deficiency is overlapped. Although multifactorial (haematinic deficiencies, shortened erythrocyte survival, low grade haemolysis, bleeding), the leading cause for CKD anaemia is the insufficient EPO production, due to decreased EPO gene expression33. The EPO gene activation in renal cells depends of microenvironmental signals and is not constitutively expressed, a possible reason why an adequate renal EPO-producing cell line is not available. In CKD, the cellular phenotype of REPCs changes to a pathologic fibrogenic state, leading to collagen production and fibrosis, which results in loss of EPO synthesis capacity.
Additionally, the uraemic environment associated with chronic inflammatory status contributes for erythrocyte survival decrease and erythropoiesis inhibition.
In fact, in CKD patients, IL-1, IL-6 and TNF-α levels are significantly amplified by the decrease in its clearance and the increase in its production34.
These pro-inflammatory cytokines contribute to anaemia and EPO resistance. Furthermore, renal failure itself contributes to inflammation with an increase in advanced glycation end products (AGE), a reduction in plasma antioxidant activity and loss of antioxidants such as zinc, selenium and vitamins C and E. This inflammatory status leads to an increase in hepcidin release by the liver, with consequent serum ferritin increase and restriction on iron availability for erythropoiesis35. The fibrosis of bone marrow induced by secondary hyperparathyroidism can exacerbate the anaemia33.
TREATMENT
Recombinant human EPO (rHu-EPO) was introduced as a treatment for CKD in 1989 (US) and 1990 (in Europe). Until then, anaemic patients were primarily controlled with blood transfusions and, to a lesser extent, with anabolic steroids, both methods with severe limitations. Regular transfusions increased the risk of infection, iron overload and anti-HLA (human leukocyte antigen) antibodies development, reducing the likelihood of a successful renal transplantation.
Steroid therapy, of limited efficacy, presented important side effects, such as hirsutism, virilization and hepatotoxicity, and was withdrawn36.
Currently, the pharmacological treatment for anaemia in CKD includes therapy with erythropoiesis stimulating agents (ESA) and supplementation with iron.
Erythropoiesis stimulating agents (ESA): all the agents able to increase, directly or indirectly, the EPO-R action. They are glycoproteins, manufactured by recombinant DNA technology, with the same biological activity as endogenous EPO.
To minimize possible risks of EPO therapy, the lowest effective dose possible should be used, seeking an increase in haemoglobin concentration near 1 g/dl per month. Full correction of anaemia with Hb > 13 g/dl may be associated with increased risk of cardiovascular and thromboembolic events30,37. The initial dose of EPO depends on the type of ESA and its administration should be considered when haemoglobin level is < 10 g/dl30. In haemodialysis patients the intravenous route is preferred, but the subcutaneous administration can substantially reduce dose requirements and is the preferred administration route in pre-dialysis, transplant or peritoneal dialysis patients, for economic and practical reasons.
In CKD and cancer patients, this treatment is associated with an increased morbidity: thrombosis and cardiovascular events (myocardial infarction, stroke and heart failure) and mortality. The risk of death or cardiovascular events is associated with a poor initial haematopoietic response, with increased ESA doses to achieve the target haemoglobin levels38.
Due to risk of tumour growth, rHu-EPO should be carefully evaluated in neoplasia setting. Erythropoietin blocks tumour cells apoptosis and potentiates angiogenesis, with increased tumour growth, metastases and reduction of the radiotherapy response39.
Hypertension and deep vein thrombosis incidence may also increase and antithrombotic prophylaxis in surgical patients treated with rHu-EPO should be considered40.
Besides CKD anaemia, rHu-EPO is currently indicated for anaemia in patients undergoing elective surgeries and cancer patients with chemotherapyinduced anaemia to reduce blood transfusions41.
The first available drugs were epoetin alfa and epoetin beta, two forms of recombinant EPO, both highly effective, but with a short duration of action (administration 3 times a week: approximate halflife of 8 hours)37. In 2001, a second-generation ESA emerged: darbepoetin alfa. It is a hyperglycosylated EPO analogue with a higher number of sialic acid residues, which improves its biological potency.
Regarding the mechanism of action, it acts as the native EPO, stimulating its receptor. Due to its high metabolic stability, it has a greater half-life time compared to conventional EPO and may be administered once every two weeks42. In 2007, the Food and Drug Administration (FDA) approved the third generation agent CERA (continuous activator EPOR)/PEG-EPO (methoxypolyethylene glycol-epoetin beta), which acts also in the EPO-R. It consists of epoetin beta PEGylated (connected to the polymer polyethylene glycol), which increases half-life: about 130 hours after the administration, by intravenous or subcutaneous routes. Thus, it can be administered every two weeks or every month, during the maintenance phase of treatment43. Another of the agents approved by FDA was peginesatide, in 2012.
This is a synthetic peptide PEGylated, with no homology to EPO, but equally capable of stimulating its receptor, initiating a similar intracellular signalling cascade (also called as EPO mimetic)44.
Its advantages include low immunogenicity and easy production, without the need for cell cultures and genetic engineering techniques and, as CERA, peginesatide could be administered monthly. However, since March 2013, this treatment can only be given to patients on dialysis due to hypersensitivity reactions associated with injection, 0.02% of them fatal45.
New erythropoietic stimulators
The new erythropoietic stimulators, summarized in Table I., search for continuous activation of EPO gene, by activating the EPO enhancer (3) through HIF stabilizers or inhibiting GATA-2, responsible for inhibiting EPO gene expression acting on its promoter.
Apart from modulation of EPO gene, the modulation of hepcidin expression, involved in the genesis of anaemia and EPO resistance, has been actively searched.
HIF stabilizers: The EPO production is controlled by transcriptional and post-transcriptional oxygendependent mechanisms. The EPO gene transcription is activated by HIF. In normoxia, HIF α chain is hydroxylated by PHD, degraded by proteasome and inactivated.
In hypoxia, α chain is not degraded and binds to β chain (constitutively expressed), leading to HIF activation and induction of EPO expression. The PHD inhibition results in HIF stabilization and EPO gene transcription. Agents that prevent HIF degradation are called HIF stabilizers. Several HIF stabilizers compounds have been studied. One of the first candidates was FG-2216, synthesized by FibroGen46. Administration of FG-2216 stabilizes HIF, promoting EPO gene transcription and increasing its synthesis. Thus, these molecules are capable of increasing endogenous EPO levels without needing an ESA. These agents have the advantage of being orally active, which represents a potential non-injectable therapy in the future. Furthermore, they are able to modulate other genes involved in erythropoiesis, in particular those related with iron utilization: transferrin, transferrin receptor, ferroportin and DMT1 (divalent metal transporter-1) and increase the iron availability.
However, there are disadvantages to be mentioned: in phase 2 clinical trial of FG-2216, one patient developed a fatal hepatic necrosis, temporally associated with HIF stabilizer administration46. In addition, several hundreds of hypoxia sensitive genes are also activated by PHD inhibition, including those involved in glucose regulation, angiogenesis, etc. One of the major concerns is related to possible vascular endothelial growth factor (VEGF) activation, which may enhance tumour growth and proliferative diabetic retinopathy. A second generation HIF stabilizer: FG-4592, presented as Roxadustat in 2013, is also orally administered and is in phase 3 trials. It acts simultaneously on erythrocytes production and iron incorporation, correcting anaemia and maintaining haemoglobin levels without needing IV iron supplementation, in pre-dialysed or dialysed patients.
The endogenous EPO production stimulation results in lower serum levels, when compared with rHu-EPO, which may be important to reduce side-effects, namely cardiovascular events, stroke or increased blood pressure, which requires introduction or intensification of antihypertensive therapy47.
GATA-2 inhibitors: The ability to positively regulate the EPO gene, through GATA-2 inhibition, has also been investigated. GATA-2 inhibits EPO gene expression, by acting on its promoter, so its inhibition can stimulate EPO gene expression and its production, enhancing erythropoiesis. There are two GATA inhibitors reported: K-7174 and K-11706. They have demonstrated to enhance EPO production and its promoter, previously suppressed by IL-1 and TNF-α. Oral administration was able to restore haemoglobin concentration, reticulocyte count, EPO levels and CFU-E (colony-forming unit erythroid) numbers.
K-11706 appears to cause a greater hypoxic induction, probably because it also stimulates HIF-1, besides GATA inhibition48. However, there is the possibility of promoting activation of other genes, like VEGF, as described with HIF stabilizers, further studies being needed.
Hepcidin modulators: Hepcidin is one of the molecules involved in anaemia genesis and endogenous EPO resistance. Thus, a suppressive therapy of hepcidin may be able to increase removal of iron from its storage and allow iron uptake from a normal diet.
It is a potential alternative to IV iron requirement, which reduces risk and side effects associated to this treatment, restoring effectiveness of oral iron therapy and increasing iron release from the reticuloendothelial system49.
Several strategies to antagonize hepcidin effect have been described. The decrease in production can be achieved by interfering in regulatory pathways.
Heparin, a widely used anticoagulant, is a potent inhibitor of hepcidin expression in liver cell lines and in mice, due to its binding capacity to BMPs. In five patients treated with low-molecular-weight heparin for deep venous thrombosis prophylaxis, serum hepcidin decreased 80 to 85%, 2 to 5 days after initiating treatment, with a concomitant increase in serum iron levels and transferrin saturation. Hepcidin inhibition and iron increase can be attributed to both anti-inflammatory activity of heparin, by IL-6 reduction, and anti-hepcidin activity through BMP sequestrations50.
In spite of the safety profile of heparin, its anticoagulant activity impairs a wider application and, for that, it was developed a non-anticoagulant heparin molecule, by glycol-split, maintaining BMPs modulation ability, which resulted in hepcidin suppression in vitro and in vivo, in mice, even in inflammatory environment, with minimal or no toxicity. Non-anticoagulant heparins have anti-hepcidin, anti-inflammatory and anti-tumour properties, representing a therapy for inflammatory anaemia in chronic diseases and cancer51.
Hepcidin regulation is modulated by BMP-6 and IL-6. This knowledge led to development of therapies directed to these targets. One example is dorsomorphin, a small molecule that inhibits BMP type I receptor and reduces hepcidin expression in inflammatory models and generalized inflammatory response itself. A dorsomorphin derivative, DLD-193189, has shown to inhibit excessive BMP signaling52 and, when used in anaemic rats for 4 weeks, produced increases in serum iron concentration and ferroportin expression, as well as an improvement in haemoglobin and haematocrit. However, besides blocking BMP pathway, it also potently inhibits VEGF and MAPK/ERK components53. Thus, contrary to what was thought, it is not a BMP specific inhibitor, representing the major challenge of these agents. Interruption of hepcidin gene activation, by IL-6, has also been proposed54.
Tocilizumab, an IL-6 neutralizing antibody, approved for rheumatoid arthritis treatment, decreases hepcidin levels in a rapid and prolonged way, with anaemia improvement in Castlemans disease.
It is a rare lymphoproliferative disorder characterized by IL-6 excessive production and elevated hepcidin serum levels, associated with microcytic hypochromic anaemia. The major complication of blocking IL-6 activity seems to be an increased risk of infections55, so this therapy should be confined to the treatment of serious diseases.
Hepcidin neutralization by an antihepcidin monoclonal antibody, tested in a mouse model of inflammatory anaemia (caused by Brucella abortus heatkilled), overcame EPO resistance. In monotherapy, it has a limited activity, but, when administered with an ESA, it demonstrated efficacy in anaemia treatment56.
Lipocalins are small extracellular proteins that exhibit a binding site with high structural plasticity.
Due to its simple structure and ability to recognize and bind to various organic compounds, they are a good class of therapeutic proteins for specific blocks.
PRS-080 anticalin is a lipocalin derivative that binds specifically to human hepcidin. In monkeys, its administration resulted in effective iron mobilization, with increased serum levels57. It is a recent therapeutic approach, needing studies for safety, tolerability and efficacy assessment. The first human clinical trial began in 2013. Another strategy consists of synthetic oligonucleotides PEGylated – spiegelmers – that bind specifically, and with high affinity, to hepcidin. This binding blocks hepcidin-induced ferroportin degradation, leading to increased serum iron concentration58.
These therapeutic agents are attractive due to high resistance to nucleases, good stability in vivo and low immunogenicity. The anti-hepcidin spiegelmer NOX-H94 was tested in monkeys with anaemia, caused by IL-6 daily injection, for 7 consecutive days. Concomitant administration of NOX-H94 decreased anaemia development59. In a phase I trial, NOX-H94 administration was safe and well tolerated, and after a single dose, there was a dose-dependent increase in serum iron and transferrin saturation. However, spiegelmers administration leads to oligonucleotides accumulation in macrophages, throughout the body, being unknown whether this chronic systemic administration results in adverse effects60. Finally, these experimental treatments for anaemia in CKD are not free of risks. Greater iron availability can promote or worsen infections, so each intervention should be properly assessed.
CONCLUSIONS
The introduction of rHu-EPO revolutionized the treatment of CKD patients, enabling a long-term safe management of anaemia, without risks associated with blood transfusions. Since then, several ESAs have appeared, characterized by lower administration frequency and better stabilization of haemoglobin concentration. New therapeutic strategies include HIF stabilizers and other non-injectable approaches, and it is expected, in a short term, the availability of an oral therapy, providing greater comfort to patient and treatment compliance.
Since iron availability is a limiting factor and its supplementation is usually necessary to ensure an adequate response to EPO, combination of different classes of agents can be an approach to equate. For example, the combination of an HIF stabilizer with a hepcidin modulator can potentiate the therapeutic effects, overcoming iron restriction.
The potential of erythropoiesis stimulation remains under investigation and it is expected that development and improvement of new therapies result in improved quality of life of patients. Further studies are needed to assess safety and long-term results, in order to apply these treatments to patients with anaemia of CKD.
References
1. Lacombe C, Mayeux P. Biology of erythropoietin. Haematologica 1998;83(8):724-732. [ Links ]
2. Jelkmann W. Molecular biology of erythropoietin. Intern Med 2004;43(8):649-659. [ Links ]
3. Paliege A, Rosenberger C, Bondke A, et al. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int 2010;77(4):312-318. [ Links ]
4. Beirao I, Moreira L, Barandela T, et al. Erythropoietin production by distal nephron in normal and familial amyloidotic adult human kidneys. Clin Nephrol 2010;74(5):327-335. [ Links ]
5. Nagai T, Yasuoka Y, Izumi Y, et al. Reevaluation of erythropoietin production by the nephron. Biochem Biophys Res Commun 2014;449(2):222-228. [ Links ]
6. Maxwell PH, Ferguson DJ, Osmond MK, et al. Expression of a homologously recombined erythopoietin-SV40 T antigen fusion gene in mouse liver: evidence for erythropoietin production by Ito cells. Blood 1994;84(6):1823-1830. [ Links ]
7. Broxmeyer HE. Erythropoietin: multiple targets, actions, and modifying influences for biological and clinical consideration. J Exp Med 2013;210(2):205-208. [ Links ]
8. Fenjves ES, Ochoa MS, Cabrera O, et al. Human, nonhuman primate, and rat pancreatic islets express erythropoietin receptors. Transplantation 2003;75(8):1356-1360. [ Links ]
9. Rankin EB, Wu C, Khatri R, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 2012;149(1):63-74.
10. Jelkmann W. Erythropoietin after a century of research: younger than ever. Eur J Haematol 2007;78(3):183-205. [ Links ]
11. Kawakami M, Sekiguchi M, Sato K, Kozaki S, Takahashi M. Erythropoietin receptormediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J Biol Chem 2001;276(42):39469-39475. [ Links ]
12. Rabie T, Marti HH. Brain protection by erythropoietin: a manifold task. Physiology (Bethesda) 2008;23:263-274. [ Links ]
13. Vesey DA, Cheung C, Pat B, Endre Z, Gobe G, Johnson DW. Erythropoietin protects against ischaemic acute renal injury. Nephrol Dial Transplant 2004;19(2):348-355. [ Links ]
14. Chateauvieux S, Grigorakaki C, Morceau F, Dicato M, Diederich M. Erythropoietin, erythropoiesis and beyond. Biochem Pharmacol 2011;82(10):1291-1303. [ Links ]
15. Calvillo L, Latini R, Kajstura J, et al. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci U S A 2003;100(8):4802-4806. [ Links ]
16. Mowat FM, Gonzalez F, Luhmann UF, et al. Endogenous erythropoietin protects neuroretinal function in ischemic retinopathy. Am J Pathol 2012;180(4):1726-1739. [ Links ]
17. Choi D, Schroer SA, Lu SY, et al. Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med 2010;207(13):2831-2842. [ Links ]
18. Jelkmann W, Bohlius J, Hallek M, Sytkowski AJ. The erythropoietin receptor in normal and cancer tissues. Crit Rev Oncol Hematol 2008;67(1):39-61. [ Links ]
19. Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 2011;1813(7):1263-1268. [ Links ]
20. Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxiainducible transcription factors, and O2-regulated gene expression. FASEB J 2002;16(10):1151-1162. [ Links ]
21. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3(10):721-732. [ Links ]
22. Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292(5516):464-468. [ Links ]
23. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001;294(5545):1337-1340. [ Links ]
24. Schödel J, Bohr D, Klanke B, et al. Factor inhibiting HIF limits the expression of hypoxia-inducible genes in podocytes and distal tubular cells. Kidney Int 2010;78(9):857-867. [ Links ]
25. Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol 2010;299(1):F1-13. [ Links ]
26. La Ferla K, Reimann C, Jelkmann W, Hellwig-Burgel T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-kappaB. FASEB J 2002;16(13):1811-1813. [ Links ]
27. Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest 2012;122(12):4635-4644. [ Links ]
28. Kemna EH, Tjalsma H, Willems HL, Swinkels DW. Hepcidin: from discovery to differential diagnosis. Haematologica 2008;93(1):90-97. [ Links ]
29. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003;102(3):783-788. [ Links ]
30. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012;2(4). [ Links ]
31. Saurabh Somvanshi NZK, Mufazzal Ahmadb. Anemia in chronic kidney disease patients. Clinical Queries Nephrology 2012;1(3):198–204. [ Links ]
32. Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001;38(4):803-812. [ Links ]
33. Macdougall IC. Anaemia and chronic renal failure. Medicine 2011;39(7):425-428. [ Links ]
34. Stenvinkel P, Ketteler M, Johnson RJ, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia–the good, the bad, and the ugly. Kidney Int 2005;67(4):1216-1233. [ Links ]
35. Vaziri ND. Oxidative stress in uremia: nature, mechanisms, and potential consequences. Semin Nephrol 2004;24(5):469-473. [ Links ]
36. Teruel JL, Marcen R, Navarro-Antolin J, Aguilera A, Fernandez-Juarez G, Ortuño J. Androgen versus erythropoietin for the treatment of anemia in hemodialyzed patients: a prospective study. J Am Soc Nephrol 1996;7(1):140-144. [ Links ]
37. Drüeke TB. Anemia treatment in patients with chronic kidney disease. N Engl J Med 2013;368(4):387-389. [ Links ]
38. Solomon SD, Uno H, Lewis EF, et al. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010;363(12):1146-1155. [ Links ]
39. Ceelen W, Boterberg T, Smeets P, et al. Recombinant human erythropoietin alpha modulates the effects of radiotherapy on colorectal cancer microvessels. Br J Cancer 2007;96(5):692-700. [ Links ]
40. Goodnough LT, Shander A. Update on erythropoiesis-stimulating agents. Best Pract Res Clin Anaesthesiol 2013;27(1):121-129. [ Links ]
41. Seidenfeld J, Piper M, Flamm C, et al. Epoetin treatment of anemia associated with cancer therapy: a systematic review and meta-analysis of controlled clinical trials. J Natl Cancer Inst 2001;93(16):1204-14. [ Links ]
42. Macdougall IC, Padhi D, Jang G. Pharmacology of darbepoetin alfa. Nephrol Dial Transplant 2007;22 Suppl 4:iv2-iv9. [ Links ]
43. Macdougall IC. Novel erythropoiesis-stimulating agents: a new era in anemia management. Clin J Am Soc Nephrol 2008;3(1):200-207. [ Links ]
44. Macdougall IC, Provenzano R, Sharma A, et al. Peginesatide for anemia in patients with chronic kidney disease not receiving dialysis. N Engl J Med 2013;368(4):320-332. [ Links ]
45. Locatelli F, Del Vecchio L. Peginesatide as a new approach for treating anemia of CKD patient: is it like a falling star? Expert Opin Pharmacother. 2013; 14(10):1277-80. [ Links ]
46. Adverse event of FG-2216 for the treatment of anemia [press release by Astellas Pharma, Inc.]. May, 2007. [ Links ]
47. Flight MH. Deal watch: AstraZeneca bets on FibroGens anaemia drug. Nature reviews Drug discovery. 2013;12(10):730. [ Links ]
48. Nakano Y, Imagawa S, Matsumoto K, et al. Oral administration of K-11706 inhibits GATA binding activity, enhances hypoxia-inducible factor 1 binding activity, and restores indicators in an in vivo mouse model of anemia of chronic disease. Blood 2004;104(13):4300-4307. [ Links ]
49. Derek S. Larson DWC. Understanding and exploiting hepcidin as an indicator of anemia due to chronic kidney disease. Kidney Res Clin Pract 2013;32(1):11-5. [ Links ]
50. Poli M, Girelli D, Campostrini N, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood 2011;117(3):997-1004. [ Links ]
51. Poli M, Asperti M, Naggi A, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood 2014;123(10):1564-1573. [ Links ]
52. Steinbicker AU, Sachidanandan C, Vonner AJ, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011;117(18):4915-4923. [ Links ]
53. Vogt J, Trainor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFß and BMP pathways. Cell Signal 2011;23(11):1831-1842. [ Links ]
54. Hashizume M, Uchiyama Y, Horai N, Tomosugi N, Mihara M. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int 2010;30(7):917-923. [ Links ]
55. Edwards CJ. IL-6 inhibition and infection: treating patients with tocilizumab. Rheumatology (Oxford) 2012;51(5):769-770. [ Links ]
56. Sasu BJ, Cooke KS, Arvedson TL, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010;115(17):3616-3624. [ Links ]
57. Fung E, Nemeth E. Manipulation of the hepcidin pathway for therapeutic purposes. Haematologica 2013;98(11):1667–1676. [ Links ]
58. Sun CC, Vaja V, Babitt JL, Lin HY. Targeting the hepcidin-ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol 2012;87(4):392-400. [ Links ]
59. Schwoebel F, van Eijk LT, Zboralski D, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 2013;121(12):2311-2315. [ Links ]
60. Bouchard PR, Hutabarat RM, Thompson KM. Discovery and development of therapeutic aptamers. Annu Rev Pharmacol Toxicol 2010;50:237-257. [ Links ]
Prof. Doutora Idalina Beirão
Department of Nephrology, Centro Hospitalar do Porto
Largo Professor Abel Salazar, 2 – 4099-001 Porto, Portugal.
E-mail: idalina.m.b@gmail.com
Conflict of interest statement: None declared.
Received for publication: 8/05/2015
Accepted: 10/05/2015

{kind=link}