










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.28 no.2 Lisboa jun. 2014
CASE REPORT
Infantile nephrotic syndrome with prominent facial dysmorphism: A possible case of Galloway-Mowat syndrome
Ayaz Ahmed1, Adeel Khalid1, Habib Qaiser1, Reema Sajjad1, Saima Qader1, Seema Hashmi1, Ali Lanewala1, Javed Iqbal Kazi2, Muhammed Mubarak2
1Departments of Pediatric Nephrology. Sindh Institute of Urology and Transplantation. Karachi, Pakistan
2Departments of Histopathology. Sindh Institute of Urology and Transplantation. Karachi, Pakistan
ABSTRACT
Galloway-Mowat syndrome is a rare hereditary disorder originally described as a triad of early-onset nephrotic syndrome, microcephaly, and hiatus hernia. Subsequent reports have expanded the clinical and pathological spectrum of the disorder. We describe a patient with this syndrome, a 14-month-old girl with infantile nephrotic syndrome having a protein to creatinine ratio of 6.1, microcephaly, low-set ears, hypertelorism, beaked nose, narrow-forehead, almond-shaped eyes, arachnodactyly, pinpoint pupils and myopia.
Renal biopsy revealed diffuse mesangial sclerosis and focal microcystic dilatation of the tubules and magnetic resonance imaging of the brain showed diffuse cortical atrophy. The above constellation of features favours the diagnosis of our case as Galloway-Mowat syndrome.
Key words: Diffuse cortical atrophy; dysmorphism; microcephaly; nephrotic syndrome; renal biopsy.
INTRODUCTION
The Galloway-Mowat syndrome (GMS) is a rare condition originally described in 1968 by Galloway and Mowat in two siblings with a triad of microcephaly, hiatus hernia and early-onset nephrotic syndrome (NS)1. It is inherited in autosomal recessive fashion whose causative genes are still unknown2-5. Since 1968, many cases with this syndrome have been reported in association with wide varieties of other abnormalities, which include anomalies of central nervous system, gut anomalies, skeletal deformities, or dysmorphic features6-9. The clinical phenotype overlaps to some extent with that of Pierson syndrome (PS), which is caused by mutations in LAMB2 gene coding for laminin s210. However, no mutations in this gene or other genes encoding functionally related proteins have been identified in GMS11,12. Only one case has been reported previously from Pakistan but without detailed renal biopsy findings or imaging of the brain12. In this report, we describe a girl with infantile onset NS, microcephaly, diffuse cortical atrophy (DCA), and prominent facial dysmorphism. Her renal biopsy showed diffuse mesangial sclerosis (DMS) and focal tubular atrophy and microcystic transformation.
CASE REPORT
A 14-month-old girl, presented with history of generalized body swelling at the age of nine months.
Initially, this swelling was very mild and limited to the periorbital area. It progressed subsequently to anasarca. She did not get any specific treatment for the ailment till she came to us. She is the second child of a consanguineous couple. Her older sister is three years old and healthy. There was no history of such illness in any family member. Mother denied any illness during her pregnancy and her natal and immediate postnatal period was not significant. An ultrasound done at seventh months of gestation was unremarkable. The anthropometric parameters at birth were not available. Her periand neonatal periodswere unremarkable.
There was no history of preceding febrile illness, rash, haematuria or seizures and she had received her scheduled immunizations with BCG, HepB, Polio, DTP and HiB. Prior to the onset of oedema, the parents reported that she was developmentally normal and was able to sit without support at nine months of age, with social smile and recognition of parents and other family members; however, over the next five months with worsening oedema, she regressed and at the time of presentation, she was very irritable, had lost her social smile and could not even hold her neck.
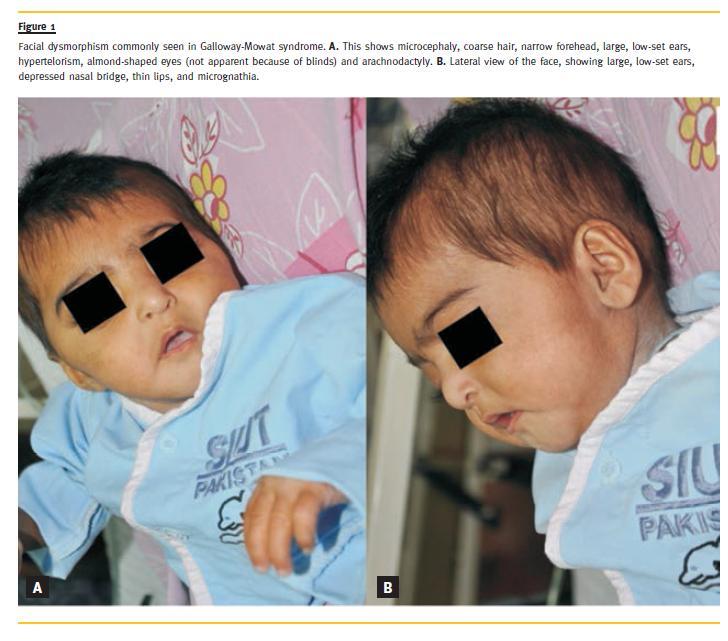
On examination, her length (64 cm) and head circumference (38 cm) was below 3rd percentile for age, while her weight (with gross anasarca) (5.7 Kg) was between 5th to 10th percentiles. Her facial features had typical beaked nose, low-set, large ears, almond-shaped eyes, narrow forehead, coarse hair, and hypertelorism (Fig. 1). Her blood pressure was 67/43 mmHg. The ophthalmologic examination by a paediatric ophthalmologist showed mega eyeballs, pin-point pupils, severe myopia and loss of foveal reflex. Her abdominal examination showed gross ascites and her fingers showed arachnodactyly (Fig. 1). Examination of the nervous system revealed no sensory or motor abnormalities. No focal deficit was elicited. The cranial nerves were intact. The rest of her physical examination was unremarkable.
Ultrasound abdomen showed moderate ascites and no hepatosplenomegaly. The right kidney measured 8.1 cm and the left, 7.8 cm.
Her baseline investigations showed serum urea, 9 mg/dl; serum creatinine, 0.46 mg/dl; low serum albumin (1.2 g/dl) with spot urinary protein to creatinine ratio of 6.1; serum total lipids of 738 mg/dl; serum triglycerides, 296 mg/dl; HDL, 22 mg/dl; LDL, 90 mg/dl; serum cholesterol, 143 mg/dl; serum total proteins, 3.4 g/dl; C-reactive protein (CRP), 3.3 ug/dl; free T4, 1.22 ng/dl; thyroid stimulating hormone (TSH), 4.62 uIU/ml, intact parathyroid hormone (iPTH), 8.7 pg/ml; C3, 0.5 g/l and C4, 0.12 g/l. Microscopic haematuria (3+) was detected but there was no glycosuria.
She was given 2mg/kg/day of prednisolone for four weeks with no significant reduction in proteinuria.
She was also given intravenous Lasix as diuretic agent, which resolved her anasarca. Nutrition consultation was obtained and her mother was encouraged to increase the quantity of weaning food in addition to breast-feeding. At four weeks after first presentation, she was clinically diagnosed as steroidresistant NS (SRNS) and the steroid dose was decreased to 1mg/kg/day every other day and the renal biopsy was performed. She was started on angiotensin converting enzyme inhibitor (ACEi), 2.5mg, once a day and started to show improvement in protein leakage.
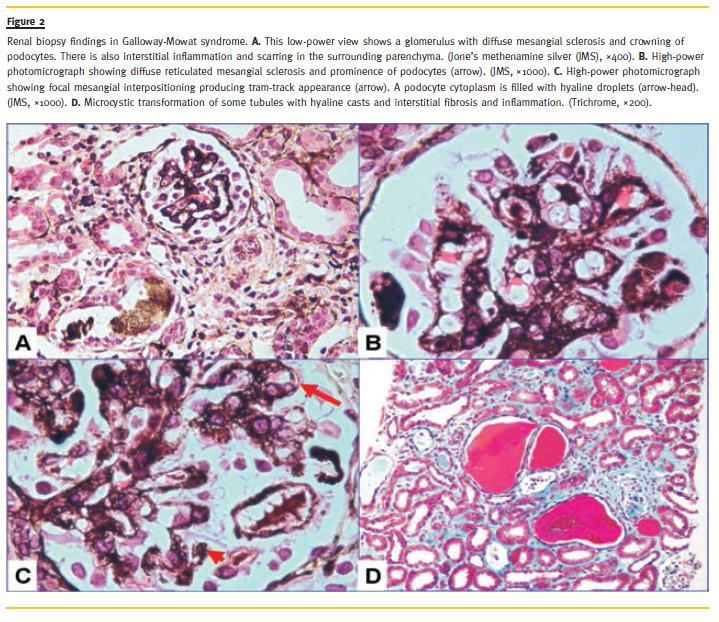
The percutaneous renal biopsy included single core of renal cortex only. Up to 50 glomeruli were included. Of these, 15 were primitive, rest show diffuse mesangial proliferation, chiefly of the matrix, and focally, the cells. Some glomeruli showed moderate mesangial proliferation with focal interpositioning into peripheral capillary loops, producing tram-track appearance. The expanded mesangial areas showed reticulated fibrosis and sclerosis. Podocytes were prominent in many glomeruli producing crown-like formation. Some podocytes also showed hyaline globules in their cytoplasm. Glomerular basement membrane (GBM) was thickened and fuzzy in outline and reduplicated in some capillary segments. Mild patchy tubular atrophy and interstitial inflammation were seen. Microcystic dilation of a few tubules with hyaline cysts was also noted (Fig. 2). Immunofluorescence was completely negative for IgG, IgA, IgM, C3 and C1q. The immunomorphological features on the renal biopsy were consistent with DMS.
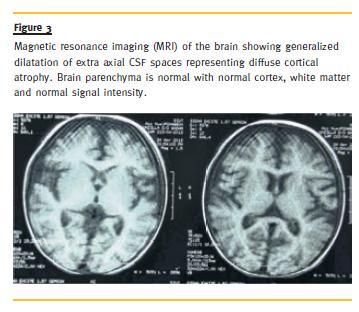
Her skeletal survey was normal. Echocardiography showed ejection fraction of 67 % and the left ventricle appeared hypertrophied. On magnetic resonance imaging (MRI) of the brain, there was generalized dilatation of extra-axial cerebrospinal fluid (CSF) spaces representing DCA (Fig. 3). Brain parenchyma including cortex, basal ganglia and white matter were unremarkable.
The colour doppler ultrasound of both legs showed no evidence of deep venous thrombosis (DVT). Genetic testing revealed no mutations in NPHS1 and NPHS 2 genes, the facility for which is available at our centre. Her chromosomal analysis could not be done due to non-availability of the test.
Meanwhile, within one week of renal biopsy, she developed a left thigh abscess, which ruptured spontaneously and large amount of pus was drained. She was empirically started on Ceftriaxone, 300 mg OD. Culture grew Pseudomonas and the antibiotic was changed to Imipenem, according to sensitivity testing and she responded dramatically.
Interestingly, following the resolution of the abscess, her oedema disappeared completely and proteinuria decreased to 2+ on dipstick testing (partial response). Her steroids were tapered and gradually withdrawn. She was discharged on ACEi.
Her renal functions remained normal throughout and the last serum creatinine was 0.34 mg/dl, which is eight months post-renal biopsy (23 months of age). Urine dipstick examination revealed 1+ protein, and no haematuria. She was oedema free.
Her weight was 6.2 Kg and height, 68 cm at the last follow-up.
Her parents were counselled on her disease and the future pregnancies. The parents were also advised for regular follow-up of the child.
DISCUSSION
Fewer than 50 cases of GMS have been reported in the English-language medical literature since the first description of the condition,by Galloway and Mowat, in 19681. Along with this, the clinical and pathological spectrum of the syndrome has also expanded over the years2-10. Although the combination of microcephaly and NS with or without hiatal hernia has been equated with GMS in the literature, the central nervous system, eye, skeletal, and renal pathology in these reported cases have been very variable. It is not entirely clear whether these represent heterogeneities within the syndrome or simply the different stages in the evolution of the disease.
There is also some phenotypic overlap with another well-known syndrome of early-onset NS and ocular anomalies, the PS. This has resulted in some confusion about the nature and distinctiveness of GMS, further aggravated by the lack of knowledge of the genes or molecules involved in this syndrome.
Although the genetic basis of PS is known, that of GMS is still unknown. The GMS is however, believed to be transmitted in an autosomal recessive pattern.
Some authors believe it to be primarily a GBM disorder with marked ultrastructural changes of GBM on electron microscopy (EM)3,8. One study found reduced expression of synaptopodin, glomerular epithelial protein-1 (GLEPP-1) and nephrin by immunohistochemistry (IHC) on renal biopsies in children with GMS. The authors suggested that this reduced expression is a non-specific secondary effect rather than a primary involvement of these genes9. Another study also analysed LAMB2, LAMA5, ITGA3, ITGB1 and ACTN4 genes, but no mutations were found in any of the above genes10. The mutational analysis was done of NPHS1 and NPHS2 genes in our case, but no pathogenic mutations were detected in these genes.
Sano et al. classified the syndrome into two types, according to the age of onset of NS and the renal histology11. The age of onset was divided into earlyonset, defined as onset within the first 3 months of life, i.e., the congenital nephrotic syndrome (CNS) and late-onset NS. This subdivision is said to be of some prognostic importance, as the prognosis in late-onset cases is somewhat favourable. Renal biopsy findings also show a wide variation of histological changes, comprising minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS) and diffuse mesangial sclerosis (DMS) with marked alterations in GBM ultrastructure, mesangial proliferative GN (MesPGN), glomerulomegaly, and microcystic renal dysplasia11. The biopsy findings also differed significantly according to the age of onset of the syndrome. The DMS was predominant in early-onset cases, while FSGS and other lesions predominated in late-onset cases of the syndrome. Our case developed NS at the age of nine months and the biopsy revealed DMS. In addition, we observed hyaline globules in hyperplastic and hypertrophic podocytes on the renal biopsy, a finding not described in literature.
The biopsy was indicated for the SRNS nature of the condition, as the syndromic presentation was subtle. It was only after the biopsy diagnosis that all the other investigations were performed to document the other abnormalities. A variety of central nervous system malformations are found in the majority of patients with this syndrome12,13. A close scrutiny of the neuroradiological and neuropathological features of the published cases reveals a fairly consistent clinical and neuropathological phenotype of the condition(13-16). An MRI of the brain in the present case revealed DCA.
It is possible to diagnose the syndrome during intra-uterine life by prenatal ultrasound in late second or early third trimester, which reveals microcephaly, intrauterine growth retardation and oligohydromnios.
These can be confirmed further by foetal ultrafast MRI17. However, other investigators failed to detect the disorder during the prenatal period by ultrasound examination only18.
The nephrotic syndrome is typically steroid-resistant and soon progresses to end-stage renal disease (ESRD). There is no specific treatment for this condition except for the supportive or symptomatic care.
Genetic counselling of the parents for future pregnancies is also warranted, as these carry a 25% risk of recurrence for the disorder. Although there was no genetic abnormality detected in our case (which is still unknown in this syndrome), we counseled the parents for possible recurrence of the syndrome in future pregnancies and the role of serial ultrasound examinations during the next pregnancy.
Overall, the long-term prognosis is not favourable, especially in early-onset cases, while the prognosis in late-onset cases, as in ours, is somewhat favourable.
Overall, the long-term prognosis is not favourable, especially in early-onset cases, while the prognosis in late-onset cases, as in ours, is somewhat favourable.
In fact, it seems that our patient has undergone spontaneous remission. She is now off the steroids and only receiving ACEi. There was no oedema and proteinuria was 1+ on dipstick on the last follow-up.
In conclusion, the early onset of NS and the phenotypical features favour a iagnosis of GMS for our patient.
We were unable to find pathogenic mutations in NPHS1 and NPHS2 genes. Renal biopsy findings exhibited a combination of glomerular and tubulo-interstitial alterations.
This case adds to the growing list of this rare syndrome in the world medical literature.
References
1. Galloway WH, Mowat AP. Congenital microcephaly with hiatus hernia and nephritic syndrome in two sibs. J Med Genet 1968;54:319-321. [ Links ]
2. Shapiro LR, Duncan PA, Farnsworth PB, Lefkowitz M. Congenital microcephaly, hiatus hernia and nephrotic syndrome: an autosomal recessive syndrome. Birth Defects Orig Artic Ser 1976;125:275-278. [ Links ]
3. Cohen AH, Turner MC. Kidney in Galloway-Mowat syndrome: clinical spectrum with description of pathology. Kidney Int 1994;455:1407-1415. [ Links ]
4. Cooperstone BG, Friedman A, Kaplan BS. Galloway- Mowat syndrome of abnormal gyral patterns and glomerulopathy. Am J Med Genet 1993;472:250-254. [ Links ]
5. Hazza I, Najada AH. Late-onset nephrotic syndrome in Galloway-Mowat syndrome: a case report. Saudi J Kidney Dis Transpl 1999;102:171-174. [ Links ]
6. Kucharczuk K, de Giorgi AM, Golden J, Zacharowicz L, van den Heuvel LP, Kaplan BS. Additional findings in Galloway-Mowat syndrome. Pediatr Nephrol 2000;145:406-409. [ Links ]
7. Shiihara T, Kato M, Kimura T, Matsunaga A, Joh K, Hayasaka K. Microcephaly, cerebellar atrophy, and focal segmental glomerulosclerosis in two brothers: a possible mild form of Galloway-Mowat syndrome. J Child Neurol 2003;182:147-149. [ Links ]
8. Lin CC, Tsai JD, Lin SP, Tzen CY, Shen EY, Shih CS. Galloway-Mowat syndrome: a glomerular basement membrane disorder? Pediatr Nephrol 2001;168:653-657. [ Links ]
9. Srivastava T, Whiting JM, Garola RE, et al. Podocyte proteins in Galloway-Mowat syndrome. Pediatr Nephrol 2001;1612:1022-1029. [ Links ]
10. Dietrich A, Matejas V, Bitzan M, et al. Analysis of genes encoding laminin beta2 and related proteins in patients with Galloway-Mowat syndrome. Pediatr Nephrol 2008;2310:1779-1786. [ Links ]
11. Sano H, Miyanoshita A, Watanabe N, et al. Microcephaly and early-onset nephritic syndrome-confusion in Galloway-Mowat syndrome. Pediatr Nephrol 1995;96:711-714. [ Links ]
12. Akhter N, Kiran S, Hafeez F. Galloway-Mowat syndrome. J Coll Physicians Surg Pak 2008;188:520-521. [ Links ]
13. Keith J, Fabian VA, Walsh P, Sinniah R, Robitaille Y Neuropathological homology in true Galloway-Mowat syndrome. J Child Neurol 2011;264:510-517. [ Links ]
14. Krishnamurthy S, Rajesh NG, Ramesh A, Zenker M. Infantile nephrotic syndrome with microcephaly and global developmental delay: The Galloway Mowat syndrome. Indian J Pediatr 2012;798:1087-1090. [ Links ]
15. Pezzella M, Yeghiazaryan NS, Veggiotti P, et al. Galloway-Mowat syndrome: an earlyonset progressive encephalopathy with intractable epilepsy associated to renal impairment. Two novel cases and review of literature. Seizure. 2010;192:132-135. [ Links ]
16. Ekstrand JJ, Friedman AL, Stafstrom CE. Galloway-Mowat syndrome: neurologic features in two sibling pairs. Pediatr Neurol 2012;472:129-132. [ Links ]
17. Chen CP, Lin SP, Liu YP, et al. Galloway-Mowat syndrome: prenatal ultrasound and perinatal magnetic resonance imaging findings. Taiwan J Obstet Gynecol 2011;502:212-216. [ Links ]
18. Kang L, Kuo PL, Lee KH, et al. Late-onset growth restriction in Galloway-Mowat syndrome: a case report. Prenat Diagn 2005;252:159-162. [ Links ]
Prof. Dr. Muhammed Mubarak
Sindh Institute of Urology and Transplantation,
Civil Hospital. Karachi, Pakistan
Email: drmubaraksiut@yahoo.com
Conflict of interest statement: None declared
Received for publication: 25/03/2014
Accepted in revised form: 25/05/2014


{kind=link}
{kind=link}