










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.27 no.2 Lisboa abr. 2013
Distal renal tubular acidosis and sensorineural deafness with mutation in the ATP6V1B1 gene
Acidose tubular renal distal e surdez neurossensorial com mutação no gene ATP6V1B1
Isabel Periquito1, Anaxore Casimiro2, Catarina E. Santo3, Cláudio DElia1, Margarida Abranches2, Isabel Castro2
1 São Bernardo Hospital, Centro Hospitalar de Setúbal, Portugal
2 Paediatric Nephrology Unit, Dona Estefânia Hospital, Centro Hospitalar de Lisboa Central, Portugal
3 Santa Maria Hospital, Centro Hospitalar de Lisboa Norte, Portugal
ABSTRACT
Distal renal tubular acidosis is a rare disorder characterized by the inability in acidification of urine, conditioning hyperchloraemic acidosis, hypokalaemia, hypercalciuria and nephrocalcinosis, which can cause growth retardation, abnormal bone metabolism and, when untreated, chronic renal failure.
Distal renal tubular acidosis and sensorineural deafness is an autosomal recessive disease caused by mutations in the gene encoding B1 subunit of H+ -ATPase (ATP6V1B1).
The authors report the cases of two sisters who presented failure to thrive, changes in ionic and acid-base balance and sensorineural deafness. A homozygous mutation in ATP6V1B1 gene was detected in both girls.
These two cases are intended to highlight the importance of an early diagnosis in this rare disease.
Keywords: Distal renal tubular acidosis, sensorineural deafness
RESUMO
A acidose tubular renal distal é uma doença rara, caracterizada pela incapacidade na acidificação da urina, condicionando acidose metabólica hiperclorémica, hipocaliémia, hipercalciúria e nefrocalcinose, o que poderá causar atraso de crescimento, alteração do metabolismo ósseo e insuficiência renal crónica.
A acidose tubular renal distal associada a surdez neurossensorial é uma doença de herança autossómica recessiva, causada por mutações do gene que codifica a subunidade B1 da H+ -ATPase (ATP6V1B1).
Os autores relatam os casos de duas irmãs que apresentaram má progressão ponderal, alterações iónicas, do equilíbrio ácido base e surdez neurossensorial. Foi detectada em ambas as crianças a mutação homozigótica no gene ATP6V1B1.
Com estes dois casos pretende-se destacar a importância de um diagnóstico precoce nesta patologia rara.
Palavras-chave: Acidose renal tubular distal, surdez neurossensorial
INTRODUCTION
Primary distal renal tubular acidosis (dRTA) is a rare entity, caused by the inability of the α intercalated cells of the cortical collecting tubules to secrete acid to the tubular lumen, resulting in severe hyperchloraemic metabolic acidosis, hypokalaemia, hypercalciuria, hypocitraturia, nephrocalcinosis and nephrolithiasis1-4.
The most frequent defects found in the paediatric population are in the H+ -ATPase, a proton pump containing several subunits encoded by different genes5. It can be sporadic or hereditary, with autosomal dominant or recessive transmission2,4,6.
The wide spectrum of clinical findings may vary from asymptomatic mild compensated acidosis to severe acidosis with growth retardation, nephrocalcinosis, urinary lithiasis, rickets and osteomalacia. If untreated the progression of nephrocalcinosis might lead to chronic renal failure1,2,3.
The authors report two cases of dRTA and sensorineural deafness in a Portuguese family, highlighting the importance of an early diagnosis and treatment of this disease.
CASE REPORTS
The cases presented are of two Caucasian siblings, of non -consanguineous parents, born and living in Portugal.
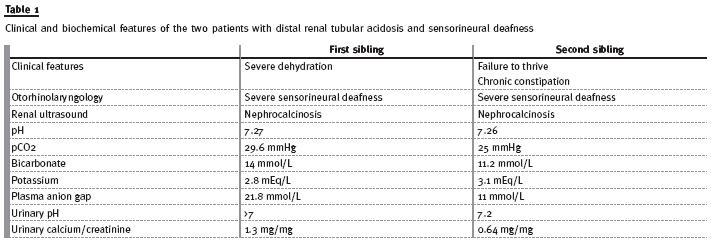
First case: third daughter of a family with four children, resulting from a full-term pregnancy, without any complications. The child was born by eutocic delivery, with a birth weight of 2570 g (10-25th percentile). She was apparently healthy until the third month of life, when the hospitalization occurred due to severe dehydration, metabolic acidosis and failure to thrive. Investigation showed: partially compensated metabolic acidosis (pH 7.27, pCO2 29.6 mmHg, bicarbonate 14 mmol/L), hypokalaemia (K+ 2.8 mEq/L), increased plasma anion gap (21.8 mmol/L), urinary pH >7 and increased urinary calcium to creatinine ratio (1.3 mg/mg). The renal ultrasound revealed alterations suggestive of nephrocalcinosis.
The child began alkali supplementation with potassium and sodium citrate and was referred to a paediatric nephrology unit for follow-up. At 32 months old, the otorhinolaryngology evaluation revealed severe sensorineural deafness, requiring auditory prosthesis. The child is now nine years old, while on oral alkali supplementation attained a well-controlled acidosis with growth improvement (weight 24Kg, 10 -25th percentile; height 132 cm, 50th percentile), and normal renal function (urea 37 mg/dL, creatinine 0,37 mg/dL).
Second case: fourth daughter of the same family, born from a poorly monitored full-term pregnancy, by eutocic delivery, with a birth weight of 2810 g (10 -25th percentile). At 14 months old, she was referred to a Paediatric appointment due to failure to thrive and chronic constipation. The laboratory results revealed a normal renal function, partially compensated metabolic acidosis (pH 7.26, pCO2 25mmHg, bicarbonate 11.2 mmol/L, base excess of -14.2 mmol/L), hypokalaemia (K+ 3.1 mEq/L), hyperchloraemia (Cl - 115 mEq/L), plasma anion gap of 11 mmol/L; increased urinary pH (pH7.2), slightly increased urinary calcium to creatinine ratio (0.64 mg/mg) and increased urinary anion gap (16 mmol/L). The renal ultrasound showed nephrocalcinosis. Taking into account the family history, she performed auditory evoked potentials test that was abnormal.
The audiometry confirmed severe bilateral, permanent sensorineural deafness. She was then referred to a paediatric nephrology unit and started oral alkali supplementation treatment with potassium and sodium citrate. She is now 4 years old, has normal growth (weight 14 kg, 10-25th percentile; height 94cm, 5-10th percentile), and normal renal function (urea 33mg/dL, creatinine 0.34 mg/dL). Follow-up of both sisters is still maintained in Paediatric Nephrology and Otorhinolaryngology departments.
The ATP6V1B1 gene sequencing was performed in both children, showing a frameshift mutation on exon 12, in gene ATP6V1B17, with both siblings being homozygous.
DISCUSSION
Distal renal tubular acidosis is a rare disorder, primary or secondary, and has a wide spectrum of clinical variability4.
Primary distal renal tubular acidosis (dRTA) may be sporadic or familial. Both autosomal dominant and autosomal recessive forms of dRTA have been recognized.
There is a subgroup of patients with dRTA, with an autosomal recessive inheritance, conferring progressive and irreversible sensorineural deafness, caused by a mutation in the gene coding a B1 subunit of the H+ -ATPase (ATP6V1B1)1-4,8. At least 15 mutations causing the disease in homozygous form have been described. Most of these mutations cause a disruption in the structure of the protein or diminish the production of the normal B1 subunit. There is a loss of expression of this gene in the human cochlea and endolymphatic sac epithelium1,9. This loss leads to a decrease in the function of the H+ -ATPase, responsible for maintaining the endolymph pH at 7.4, causing irreversible hair cell damage, in the organ of Corti5. The clinical manifestations, aside from deafness, are similar to those of the other types of dRTA1,2. There are other mutations, namely the ATP6V0A4 encoding the a4 subunit and the ATP6N1B encoding the 116 -kD subunit of the H+ -ATPase, also associated with deafness, particularly after the second decade of life8,10,11. The correlation between the genetic mutation and deafness is not absolutely clear, since there are some patients with the ATP6V1B1, who do not present the same typical severe early-onset sensorineural deafness5.
Failure to thrive, a relatively common occurrence in the paediatric age, was the initial symptom in both cases, as previously described in other published cases12, therefore not contributing significantly for a swift diagnosis. In the first case, dehydration was the motive for an early evaluation in the emergency department, favouring earlier diagnosis of the disease.
Laboratory results confirmed that both children have a rare hereditary form of dRTA, of recessive autosomal inheritance. Taking into account that the parents are probably heterozygous for the same mutation, there is a 25% risk of recurrence in future gestations.
The diagnosis of this disease requires a high degree of suspicion, since the symptoms are usually non-specific and highly variable. Initially one might only find fatigue, constipation or myalgia, due to the neuromuscular effect of hypokalaemia5,12 or, on the contrary, the first manifestation may be dehydration with severe metabolic acidosis.
CONCLUSION
The authors intend with these two cases to highlight the importance of the family history and a high degree of suspicion for the diagnosis of dRTA.
Delaying the diagnosis of this pathology has important implications in the ionic balance, growth5 and development of a child, as well as the progression to nephrocalcinosis and, eventually, chronic renal failure11. Most complications are reduced with adequate treatment, however, nephrocalcinosis will not regress despite the correction of acidosis and therapy with diuretics5,12. Patients with ATP6V1B1 mutations, as in our cases, are more likely to develop progressive loss of kidney function, probably secondary to nephrocalcinosis13.
table I
The authors would also like to stress the importance of a careful follow-up of these patients in specialized centres, with Paediatric Nephrology, for a well-controlled alkali supplementation and monitoring of glomerular -filtration rates, and otorhinolaryngology, for educational support and speech therapy to help improving deafness-related disabilities.
A genetic evaluation is also of great importance, not only to confirm the suspected diagnosis, but also for genetic counselling.
References
1. Sethi SK, Singh N, Gil H, Bagga A. Genetic studies in a family with distal renal tubular acidosis and sensorineural deafness. Indian Pediatr 2009;46(5):425-427 [ Links ]
2. López Hidalgo R, Polo Moyano A, Manjón Rodríguez M, Cerezo Morales S. Acidosis tubular renal distal con sordera neurosensorial. Evolución clínica tras 30 años de seguimiento. Nefrología 2009;29(5):499-500 [ Links ]
3. Gil H, Santos F, García E, et al. Distal RTA with nerve deafness: clinical spectrum and mutational analysis in five children. Pediatr Nephrol 2007;22(6):825-828 [ Links ]
4. Karet FE. Inherited distal renal tubular acidosis. J Am Soc Nephrol 2002;13(8):2178-2184 [ Links ]
5. Chan J, Santos F. Renal tubular acidosis in childhood. World J Pediatr 2007:3:92-97 [ Links ]
6. Vargas-Poussou R, Houillier P, Le Pottier N, et al. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol 2006;17(5):1437–1443 [ Links ]
7. Karet F, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+ -ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 1999;21(1):84-90 [ Links ]
8. Stover EH, Borthwick KJ, Bavalia C, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 2002; 39(11):796-803 [ Links ]
9. Feldman M, Prikis M, Athanasiou Y, Elia A, Pierides A, Deltas CC. Molecular investigation and long-term clinical progress in Greek Cypriot families with recessive distal renal tubular acidosis and sensorineural deafness due to mutations in the ATP6V1B1 gene. Clin Genet 2006; 69(2):135–144 [ Links ]
10. Laing CM, Toye AM, Capasso G, Unwin RJ. Renal tubular acidosis: developments in our understanding of the molecular basis. Int J Biochem Cell Biol 2005 Jun; 37(6):1151-1161 [ Links ]
11. Rodriguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 2002;13(8):2160-2170 [ Links ]
12. Chan JC, Scheinman JI, Roth KS. Consultation with the specialist: renal tubular acidosis. Pediatr Rev 2001;22(8):277-287 [ Links ]
13. Vivante A, Lotan D, Pode-Shakked N, et al. Familial Autosomal recessive renal tubular acidosis: Importance of early diagnosis. Nephron Physiol 2011;119(3):31-39 [ Links ]
Isabel Rosário Periquito
Centro Hospitalar de Setúbal
Rua Camilo Castelo Branco
2910-446 Setúbal, Portugal
E-mail: isabelperiquito@gmail.com
Conflict of interest statement: None declared.
Received for publication: 15/01/2013
Accepted in revised form: 25/03/2013


{kind=link}