










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.26 no.4 Lisboa out. 2012
NSAID-related renal tubular dysfunction, an old new conundrum
Wael Lateef Jabur
NMC Specialty Hospital. Dubai-UAE
ABSTRACT
Aim.To report on ten patients who presented withacute kidney injury secondary to nonsteroidal anti-inflammatory drug administration. Renal tubular function test was consistent with acute tubular necrosis; however kidney biopsy in two of them revealed a normal ultrastructural finding, which along with the atypical clinical course refuted the suggested diagnosis.
The constellation of features pointed to a clear reversible tubular dysfunction in the absence of tubular necrosis, highlighting a peculiar mechanism that might provoke acute kidney injury, involving the inhibition of the prostaglandin, the integral mediator of the tubular function, in addition to its role in maintaining the perfusion of the glomeruli and supporting the function of the macula densa. Conclusively a potential mechanism, apart from acute tubular necrosis and haemodynamic renal failure, was considered in this cohort of patients to explain nonsteroidal anti-inflammatory drug-related AKI, with an elaborate discussion of the particular findings of renal tubular function test, focusing on the probable physiological aberration that might culminate in nonsteroidal anti-inflammatory drug-related nephropathy.
Patients and Methods. Ten patients attended the NMC specialty hospital in Dubai from 2006 through 2011 with acute kidney injury secondary to nonsteroidal anti-inflammatory drug use, which developed within 2-5 days. The main complaints were abdominal pain, nausea and vomiting. Blood pressure was elevated in all of them. Renal tubular function test revealed characteristic findings in all of them. Kidney biopsy was performed in two patients.
Results. Investigations revealed prominent renal tubular dysfunction suggestive of acute tubular necrosis, with high sodium fractional excretion of 3-5 %, high uric acid fractional excretion of 12-13 %, urine osmolality to plasma osmolality ratio of 1, urine creatinin to plasma creatinin ratio of 16, and urine specific gravity of 1.010-1.020. However kidney biopsy in two of them showed normal renal tubular architecture, which in combination with the short-term, reversible course of the kidney injury in the rest of the patients coincided with nonstructural acute renal tubular dysfunction. Management was supportive.
Most of them improved within 5-7 days and two of them improved within 40-45 days.
Conclusion. This report highlights that renal tubular dysfunction might be the consistent primary lesion in some of the cases of nonsteroidal anti-inflammatory drug-related acute kidney injury, notwithstanding that acute tubular necrosis and interstitial involvement were evidently excluded. This might be a new explanation for the mechanism of renal failure in patients with otherwise normal renal function.
Key-Words: Acute kidney injury (AKI); acute renal tubular dysfunction; nonsteroidal anti-inflammatory drugs (NSAID).
INTRODUCTION
Nonsteroidal anti-inflammatory drug (NSAID)-related acute kidney injury (AKI) is an apparent risk when there is compromised kidney function and enormous dependence on the prostaglandin system.
Despite the fact that it is an important vasodilator and potent antagonist to the major renal vasoconstrictors, namely the angiotensin II and the endothelin, the prostaglandin system is relatively inactive in the circumstances of normal glomerular perfusion and haemodynamics.
Considering these facts, the precipitation of AKI in otherwise normal kidneys by the inadvertent use of NSAID is apparently unusual. I report on ten patients with normal prior kidney function diagnosed with AKI secondary to NSAID usage and discuss some of their unusual findings and their possible explanations.
PATIENTS AND METHODS
Ten patients attended the NMC specialty hospital in Dubai from 2006 through 2011 with a diagnosis of AKI. Age ranged between 20-40 years old, and there were eight males and two females. All were asymptomatic until a few days before admission.
They did not report any past history of significance.
Their presentation was marked by initial acute painful illness (acute flu-like illness and joint or muscle pain) for which over-the-counter pills including a wide range of NSAIDs were the main therapy (Table I).
They did not, however, improve, and new symptoms of nausea, vomiting and abdominal pain emerged, necessitating their admission to the hospital with an exclusion diagnosis of AKI secondary to the use of NSAID. This developed within 2-5 days of the initial illness. Blood pressure was elevated in all of them.
Clinical and investigational features of the ten patients in the study
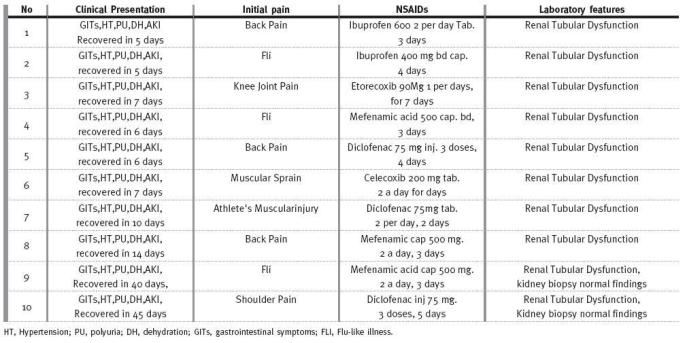
RESULTS
The presentations of the ten patients were similar in terms of abruptness of deterioration of renal function, lack of remarkable history of kidney disease, scarcity of the clinical findings, the normal ultrasonic echogenisity and renal size, and the ultimate recovery of the kidney function in all of them (Table I). Thorough investigations including antinuclear antibody, antineutrophil cytoplasmic antibodies, antiglomerular basement membrane antibody, C3, C4 and total complements were unremarkable in all of the ten patients. Kidney biopsy was performed in two patients because of the unusually extended course of AKI for 40-45 days. This revealed normal histology, precluding the presumed diagnosis of acute tubular necrosis (ATN). The severity of renal impairment was comparable in each, and serum creatinine rose to 3-4 mg/dL. Urine output was of normal to high volume (3-4 litres/day). Hypertension of new onset was reported in all, and it was of accelerated phase in one of them. Hyperuricaemia of 9-11.5 mg/dL was reported in all patients. Urine analysis was unremarkable for protein or active sediments.
Renal tubular function indices were characteristically featuring peculiar derangements in all, including Na fractional excretion of 3-5 %, total urinary Na of 80-90 mmol/l and fractional uric acid excretion of 13-14 %. Urinary creatinin to serum creatinin ratio was 15-17. Urine specific gravity ranged from 1.010 to 1.020. Serum sodium, potassium and chloride were within normal limits. Management consisted of isotonic intravenous fluids, anti-emetics and antihypertensives in the form of calcium channel blockers and beta blockers. In all patients renal function improved gradually over a period of 5-45 days (Table I).
DISCUSSION
AKI secondary to NSAID is of two aetiologies1. It is either structural due to acute interstitial nephritis, in which eosinophiluria and minimal-change-diseaserelated nephrotic syndrome are common companions, or secondary to haemodynamically-mediated renal hypoperfusion. Both conditions might be provoked by the interruption of the renal prostaglandin system.
The gastrointestinal symptoms seen in all the patients in association with AKI were attributed to NSAID consumption.
Despite renal tubular function indices being consistent with ATN (high fractional excretion of Na and uric acid, 3-5% and 13-14% respectively, and the polyuria), which might be consequent to profound renal hypoperfusion, I am inclined to consider the underlying pathology as a functional impairment of renal function. The reasons for this are the absence of the well-defined three-phase sequence of initiation, maintenance and recovery that is characteristic of ATN, and the short-term and promptly reversible renal course of less than seven days (Table I) in eight out of ten patients.
No kidney biopsy was contemplated in any of the early recovering patients to prove this assumption, however, and it was evidently elucidated in two of the patients who underwent kidney biopsy, with a primary presumption that ATN was the most probable aetiology of their AKI, due to the comparatively prolonged course of 40-45 days. Interestingly, kidney biopsies revealed normal renal histology, as well as normal ultrastructural findings by electron microscopy study of the renal tubules. The widely recognized mechanism behind haemodynamically mediated AKI2 of the inhibition of the prostaglandins PGE2 and PGI2 usually results in afferent glomerular arteriolar vasoconstriction, leading to glomerular hypoperfusion and declining glomerular filtration rate, denoting a prerenal aetiology of AKI.
Interestingly, NSAID administration is usually associated with sodium, chloride and water retention, because of the current belief that the prostaglandins are inhibiting the reabsorption of NaCl and antagonizing the effect of the vasopressin hormone1. This mechanism would have been more pronounced in compromised kidney function, and the kidneys are dependent on the prostaglandin system to maintain normal glomerular filtration rate, however2. The normal kidneys otherwise would have not been affected by inhibition of the prostaglandin system because of its low rate of production.
Curiously, patients histories reported, on the contrary, normal prior kidney function, and their investigations revealed prominent renal tubular dysfunction indices and not that of the widely recognized pre-renal indices typical of NSAID-related AKI. Considering the similar peculiar clinical and investigational presentation in these patients (Table I), I incline towards the particular aetiology of reversible functional tubular damage incurred by the acute effect of the NSAID. Although the occurrence of AKI in normal healthy individuals secondary to NSAID has been reported in children3 and adults, this presentation would explicitly rival the currently held theory of effects on the cortical and medullar levels4. That said, the association of AKI with functional renal tubular impairment secondary to NSAIDs is not yet widely recognised, and I think the concurrent presentation is more consistent with NSAID-related AKI, since the prostaglandins are vastly involved in the tubular function. It is also well-known as a modulator of the macula densa-secreted renin in response to sodium chloride concentration in the ascending loop of Henle5. In addition, the up regulation and overexpression of it in specific tubular disorders such as Bartter syndrome would highlight its pivotal role6.
Although the underlying mechanism remains to be elucidated, it is obviously an aberration of the normal prostaglandin function both in normal and compromised kidney function status. It might highlight the potential of another underlying mechanisms being involved in NSAID-related AKI, a primarily functional renal tubular impairment and secondarily compromised glomerular filtration function. The tremendous natriuresis observed in all the patients despite the prominent dehydration they presented with might have been the key to explain the renal function impairment.
While no plasma renin activity or plasma aldosterone level measurements were undertaken, it hints at the generalised renal tubular failure of reabsorbing NaCl, and salt-losing nephropathy. This might be explained to a greater extent by the failure of the macula densa to maintain the intrarenal renin necessary for the reabsorption of the NaCl, either by the action of angiotensin II on the proximal tubule, or through the renin-angiotensin-aldosteron system operating on the distal tubule.
On the other hand it might highlight the impairment of the tubulo-glomerular feedback mechanism7, which would otherwise be operational to adjust the excessive delivery of NaCl to the distal nephron.
Although adenosine is widely recognised as the mediator of the tubuloglomerular feedback mechanism8, this presentation might foster the probable role of prostaglandin in this mechanism, and interestingly would bolster the suggestion of the impaired function of macula densa5, since the tubuloglomerular feedback mechanism is dependent to a greater extent on the function of macula densa and the presence of angiotensin II.
The large volume diuresis of reduced specific gravity urine is consistent with failure of distal nephron to conserve water, and since it was a functional impairment, it is mostly related to the failure of the vasopressin hormone-mediated absorption of water, a finding that cannot be explained by prostaglandin inhibition. It might be secondary to loss of the medullary hypertonisity incurred by massive natriuresis and failure of the proximal nephron to reabsorb NaCl, and/or because of excessive delivery of NaCl to the distal nephron.
Conversely, failure of the macula densa-mediated renin release would hamper the compensatory haemodynamic mechanism that operates to maintain intraglomerular pressure and consequently the filtration function. Failure of NaCl reclaiming and glomerular haemodynamic response would result in a relative hypovolaemic state and renal hypoperfusion What is intriguing is the hypertension reported in all patients. It would negate our belief of having a hypovolaemic, hyporeninimic state. However it might be highlighting the effect of systemic inhibition of prostaglandin in triggering hypertension9.
Summing up the whole deliberations would propose that those patients showed a constellation of syndromes, including AKI, hypertension, and saltlosing nephropathy, that were attributed mainly to NSAIDs, as the sole aetiology, for the following reasons.
Firstly, by exclusion of other possible aetiologies (including kidney biopsy in two of the patients).
Secondly, the lack of significant past medical history, the short-term and reversible course of AKI in most of the cases and most importantly the peculiar renal tubular dysfunction in all the patients which was not reflective of other possible causes. Dehydration and hypovolaemia, as highlighted earlier, was arguably believed to be the main culprit for AKI, provoked by the tremendous renal tubular dysfunction causing salt and water loss, which in turn was invoked by the initial administration of NSAIDs.
CONCLUSION
We point out a few salient issues. Firstly, the occurrence of AKI secondary to NSAID use in patients with otherwise normal kidney function, who yet might be genetically predisposed. Secondly the peculiar tubular dysfunction that might implement functional renal failure. Thirdly that the probable underlying pathogenic mechanism is failure of the macula densa to secrete renin, and failure of tubuloglomerular feedback mechanism, both of which will lead to massive natriuresis and failure of intraglomerular haemodynamics, leading to renal failure as an aftermath of the tremendous tubular functional defect.
This might be a new explanation for renal failure in NSAID-related AKI in patients with otherwise normal renal function. The main caveats of this study are the small number of the patients enrolled and the need for more sophisticated measurements of the plasma renin and urine prostaglandin to foster the theory.
References
1. Latha V. Cyclooxygenase-2 Inhibitor-associated acute renal failure. Pharmacotherapy 2002;22:1317-1321 [ Links ]
2. Perazella MA, Eras J. Are COX-2 selective inhibitors nephrotoxic. Am J Kidney Dis 2000;35:937-40 [ Links ]
3. Krause I, Cleper R, Eisenstein B, et al. Acute renal failure associated with non-steroidal anti-inflammatory drugs in healthy children. Pediatric Nephrol 2005;20:1295-1298 [ Links ]
4. Hao CM, Breyer MD. Physiological Regulation of Prostaglandins in the Kidney. Annu Rev Physiol 2008;70:357-377 [ Links ]
5. Harris RC, Wang JL, Cheng HF, et al. Prostaglandins in macula densa function. Kidney Int 1998;54:s49-s52 [ Links ]
6. Kleta R, Basoglu C, Kuwertz-Droking E. New Treatment Options for Bartters syndrome. N Engl J Med 2000;343:661-662 [ Links ]
7. Kmlosi P, Bell PD, Zhang ZR. Tubuloglomerular feedback mechanisms in nephron segments beyond the macula densa. Curr Opin Nephrol Hypertens 2009;18:57-62 [ Links ]
8. Schnermann J, Levine DZ. Paracrine Factors in Tubuloglomerular Feedback. Adenosine, ATP and Nitric Oxide. Annu Rev Physiol 2003;65:501-529 [ Links ]
9. Colina-Chourio JA, Odoy-Godoy N, Avilahernandez RM. Role of Prostaglandins in hypertension. J Hum Hypertens 2000;14(Supp 1):S16-S19 [ Links ]
Wael Lateef Jabur MD, FACP
Consultant Nephrologist
NMC Specialty Hospital
Dubai-UAE
E-mail: drwaellatif@hotmail.com
Conflict of interest statement.None declared.
Received for publication: 28/08/2012
Accepted in revised form: 12/11/2012

