










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.26 no.2 Lisboa abr. 2012
Phosphate balance in chronic kidney disease: the chicken or the egg?
Teresa Adragão1, João M. Frazão2
1 Nephrology Department, Hospital de Santa Cruz. Carnaxide, Portugal.
2Nephrology Research and Development Unit and School of Medicine, University of Porto; and Nephrology Department, Hospital S. João, Porto, Portugal.
ABSTRACT
In chronic kidney disease patients there are three main stimuli for parathyroid hormone (PTH) secretion by the chief cell in the parathyroid glands: hypocalcaemia, low 1,25(OH)2D3 levels and hyperphosphataemia.
FGF23 is a regulator of phosphate and vitamin D metabolism. The discovery of FGF23 actions enlightened our understanding of the development of secondary hyperparathyroidism in CKD patients. The main systemic factors that stimulate FGF23 secretion by the osteocyte in the bone appear to be phosphate load and 1,25(OH)2D3. In the kidney, FGF23 decreases the number of Na/Pi co-transporters IIa and IIc in the tubular cell and promotes phosphaturia. FGF23 also reduces 1,25(OH)2D3 levels by inhibiting, in the kidney, its production by 1-alpha-hydroxylase and stimulating its degradation by 24-hydroxylase. Increase in FGF23 levels has been described in early 2 and 3 CKD stages preceding the decrease of 1,25(OH)2D3 levels and hyperphosphatemia. In this sequence of events, increase of FGF23 in CKD patients seems to be a novel mechanism for the early decline of 1,25(OH)2D3 levels observed in these patients. It was hypothesised that klotho deficiency creates a tissue resistance to FGF23 which is responsible for the increase of FGF23 levels. Reduced renal expression of klotho has been demonstrated in CKD patients preceding FGF23 increase. Chronic kidney disease may be considered a state of klotho deficiency with increase of FGF23 levels. Klotho deficiency may be the initial alteration for the development of phosphate retention and secondary hyperparathyroidism in CKD patients. In this article we review the classic and new pathways involved in the development of secondary hyperparathyroidism in chronic kidney disease and the subsequent actions ensuing from this knowledge.
It is possible that, in 3 and 4 CKD stages, an early therapeutic intervention consisting of a low phosphate diet and/or phosphate binders, even in the presence of normophosphataemia, might retard the development of secondary hyperparathyroidism.
Key-Words: Bone-kidney-parathyroid endocrine axis; chronic kidney disease; FGF23; Klotho; phosphate balance.
STIMULI FOR PARATHYROID HORMONE SYNTHESIS AND SECRETION WITH THE CONSEQUENT DEVELOPMENT OF SECONDARY HYPERPARATHYROIDISM IN CHRONIC KIDNEY DISEASE
In chronic kidney disease (CKD) patients there are three main stimuli for parathyroid hormone (PTH) secretion by the chief cell in the parathyroid glands: hypocalcaemia, low 1,25(OH)2D3 levels and hyperphosphataemia1 (Fig.1). Low 1,25(OH)2D3 levels and low calcium levels directly stimulate the parathyroid cell through their action on specific receptors, the vitamin D receptor2 in the nucleus and the calcium receptor3 in the cell membrane, respectively. A receptor for phosphate has not yet been identified in the parathyroid cell, but a direct effect of hyperphosphataemia on PTH secretion, independent of its effect on calcium and 1,25(OH)2D3, has already been demonstrated4-6; 1,25(OH)2D3 decreases PTH gene transcription7 and calcium and phosphate regulate the PTH gene post-transcriptionally8.
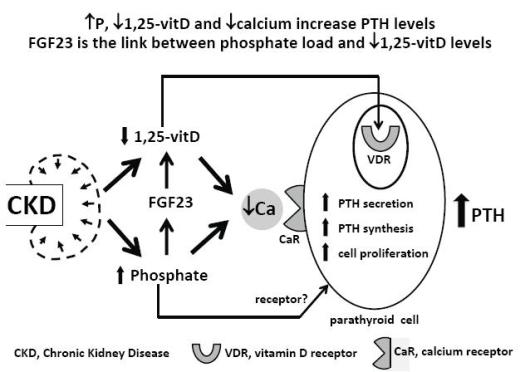
Development of secondary hyperparathyroidism in CKD
In the normal parathyroid gland few cells proliferate. In secondary hyperparathyroidism there is an increase in parathyroid cell number, in PTH gene expression and secretion. Hypocalcaemia, hyperphosphataemia and uraemia lead to parathyroid cell proliferation9.
CALCIUM, PHOSPHATE AND 1,25(OH)2D3 LEVELS DURING CKD PROGRESSION
1.Hyperphosphataemia contributes to the development of secondary hyperparathyroidism
The trade-off hypothesis has conferred on phosphate a pivotal role in the development of secondary hyperparathyroidism10 and hyperphosphataemia has been considered the primordial stimulus for the development of this pathological condition11. The observation that hyperphosphataemia is only present in late 4 and 5 CKD stages12,13 has cast doubts on phosphates alleged role in stimulating PTH secretion in early CKD stages. However, this imaginative hypothesis holds true when applied to more advanced CKD stages. The mechanisms commonly considered as explaining the development of hyperparathyroidism in consequence of hyperphosphataemia are the phosphorus-induced decrease in 1,25(OH)2D3 levels, the phosphorus-induced hypocalcaemia and the direct independent effect of phosphorus on parathyroid cell function11.
2. 1 ,25(OH)2D3 deficiency contributes to the development of secondary hyperparathyroidism
The hypothesis that vitamin D plays the primordial role in the progression of secondary hyperparathyroidism has also been raised14. This hypothesis has been strengthened by more recent studies demonstrating that reductions of 1,25(OH)2D3 levels appear in early CKD stages and precede the development of hyperphosphataemia15,16.
3. H ypocalcaemia contributes to the development of secondary hyperparathyroidism
The development of hypocalcaemia is an event that occurs in late CKD stages12,13 and has been explained by several factors, such as the lower renal tubular reabsorption from the failing kidney, the lower intestinal absorption of calcium in relation with low 1,25(OH)2D3 levels and the lower release of Ca from bone in relation to hyperphosphataemia17.
Inappropriate postprandial calciuria with episodic relative hypocalcaemia and increase in PTH levels in 3 and 4 CKD stages is a new proposed mechanism for the development of secondary hyperparathyroidism driven by hypocalcaemia in early CKD stages18.
FIBROBLAST GROWTH FACTOR 23
Fibroblast growth factors family members are now defined as humoral factors which have in common a three-dimensional β-trefoil structure. To date, twenty-two human fibroblast growth factors have been identified (1 to 14 and 16 to 23) and grouped into seven subfamilies. Fibroblast growth factor (FGF) 23 was identified as the last member of the FGF superfamily and belongs to the FGF19 subfamily.
FGFs execute their biological action by binding to an FGF receptor (FGFR) with an extracellular domain, a single-pass transmembrane domain and an intracellular domain responsible for a tyrosine kinase activity19. Contrary to the other FGFs which act in a paracrine way, FGF19 subfamily members achieve their activities in an endocrine fashion. In paracrine FGFs, stable FGF-FGFR binding is regulated by heparin and heparan sulphate20. In the FGF19 subfamily heparin or heparan sulphate have a poor ability to promote binding to FGFR, and FGF19 subfamily members require the presence of Klotho or beta-Klotho in their target tissues21. Membrane Klotho forms a complex with FGFR and functions as an obligate co-receptor for FGF2321. The restricted tissular expression of Klotho proteins also contributes to the endocrine behavior of this subfamily by limiting the signalling of these ligands to the specific tissues21. Membrane Klotho has been identified in kidney, in choroid plexus in brain and in parathyroid glands.
The FGFs are now considered to play substantial roles in development, angiogenesis, haematopoiesis and tumorigenesis. FGF19 subfamily members regulate diverse physiological processes uncommon to classical FGFs21, namely bile acid homeostasis (FGF19), glucose and lipid metabolism (FGF21) and phosphate and vitamin D homeostasis (FGF23).
FGF23 AND THE BONE-KIDNEYPARATHYROID ENDOCRINE AXIS: A LINK BETWEEN PHOSPHATE LOAD AND LOW VITAMIN D LEVELS
FGF23 is a regulator of phosphate and vitamin D metabolism. It is a 32-kDa protein with 251 amino acids that is secreted mainly by osteocytes in bone22.
The discovery of FGF23 actions enlightened our understanding of the development of secondary hyperparathyroidism in CKD patients. The main systemic factors that stimulate FGF23 secretion by the osteocyte in the bone appear to be phosphate load23 and 1,25(OH)2D324. In the kidney, FGF23 decreases the number of Na/Pi co-transporters IIa and IIc in the tubular cell and promotes phosphaturia25,26.
FGF23 also reduces 1,25(OH)2D3 levels by inhibiting, in the kidney, its production by 1-alpha-hydroxylase25,26 and stimulating its degradation by 24-hydroxylase26.
In parathyroid glands, FGF23 suppresses production and secretion of PTH. Suppression of PTH contributes to the reduction of 1,25(OH)2D3 levels.
Klotho is much more abundant in distal convoluted tubules than in proximal tubules. It is not known whether FGF23 acts directly or indirectly in the proximal tubule, promoting phosphaturia and inhibiting 1,25(OH)2D327.
Increase in FGF23 levels has been described in early 2 and 3 CKD stages28-30 preceding the decrease of 1,25(OH)2D3 levels and hyperphosphataemia30,31.
In this sequence of events, increase of FGF23 in CKD patients seems to be a novel mechanism for the early decline of 1,25(OH)2D3 levels observed in these patients32. Increased phosphate load stimulates the synthesis of FGF2325 and FGF23 promotes phosphaturia and maintains phosphate levels between normal ranges. The price to maintain normal phosphate levels is the decrease in 1,25(OH)2D3 levels with the subsequent stimulation of PTH synthesis. In this new paradigm FGF23 is a determinant player in the pathophysiology of secondary hyperparathyroidism in early CKD stages32.
Phosphate loading in mice increases FGF23 levels25, but the data in humans are conflicting. Hyperphosphataemia induced by intravenous phosphorus infusion was not associated with increase in FGF23 levels33. After an oral phosphate load, an increase in the fractional excretion of phosphate was observed in early CKD patients18 and healthy volunteers34 but FGF23 levels did not increase immediately after the oral phosphate load. However, 8 hours after phosphate ingestion, a 20% increase of FGF23 levels was observed in healthy volunteers34.
The mechanism by which phosphate regulates FGF23 production is still unknown. A phosphate sensor that controls FGF23 production has not been identified and extracellular phosphate has not been shown to regulate FGF23 gene transcription in osteoblast cultures24, raising the possibility that phosphate effects on FGF23 production might be indirect35.
In theory, a low phosphate diet and/or the administration of phosphate binders in CKD stages3 and 4, even with normophosphataemia, may prevent the development of mineral abnormalities associated with secondary hyperparathyroidism36. Some of the beneficial effects of phosphate restriction in early CKD stages were described more than 25 years ago37,38. It was found that a restriction in the dietary intake of phosphorus in patients with moderate renal insufficiency was associated with a decrease in fractional excretion of phosphate37, increase in 1,25(OH)2D3 levels37,38 and a decrease in iPTH levels37,38.
These findings, due to the recent discovery of FGF23 actions, may now be interpreted in a new light.
The effect of phosphate binders in preventing gastrointestinal phosphate absorption and phosphate load has been evaluated in two pilot studies in normophosphataemic CKD stages 3 and 4 patients39,40. During a 6-wk period followed by a 2-wk washout period, calcium acetate and sevelamer treatment were associated with a decrease in PTH levels and urinary phosphate; in the sevelamer group but not in the calcium acetate group there was also a decrease in FGF23 levels after the 4th week of treatment39. This latter observation of a different effect of calcium acetate and sevelamer on FGF23 levels still is not fully understandable in the light of present knowledge, but some authors suggest that might be the consequence of a direct effect of calcium load on osteocytes. This study raises the interesting possibility of controlling the mineral metabolism disturbances and secondary hyperparathyroidism with the early control of phosphate loading.
In a very short study with only a 2-wk duration40, lanthanum carbonate and dietary phosphate restriction lowered urinary phosphate excretion without lowering FGF23.
These interesting preliminary results need to be confirmed in appropriate clinical trials.
The source of protein has also a different effect on phosphorus homeostasis36. The diurnal variation of phosphate levels, fractional excretion of phosphate and iPTH levels were consistently lower in a vegetarian diet than a meat diet with similar protein and phosphate content36. The source of phosphate should, therefore, be included in the dietary counseling for CKD patients.
FGF23 AND CLINICAL OUTCOMES
FGF23 is a more robust predictor of adverse outcomes in CKD patients than serum phosphate levels31.
Increased levels of FGF23 have been associated with mortality in incident41 and prevalent42 haemodialysis patients and in patients with stable coronary disease, without end-stage renal disease43. Increased risk of cardiovascular events has been also predicted by high FGF23 levels in CKD patients not on dialysis44.
FGF23 increase has been associated with left ventricular hypertrophy45, left ventricular mass, severe vascular calcification46 and with increased risk of CKD progression47. However, all these observational studies can only demonstrate an association effect between high FGF23 levels and adverse clinical outcomes, and another type of evidence, such as randomised clinical trials, is needed to demonstrate that high FGF23 levels are the cause of these adverse outcomes.
Another intriguing aspect of the possible causal relationship between FGF23 and cardiovascular events is the fact that although klotho is needed as a co-factor for FGF23 klotho is not expressed in the cardiovascular system, and there is a klotho deficiency in CKD patients48. It was hypothesised that very high FGF23 levels, independent of klotho collaboration, may activate other FGF receptors31 but klotho deficiency also contributes directly to vascular calcification48. A recent study has definitely demonstrated a direct contribution of FGF23, independent of klotho, in the development of left ventricular hypertrophy in mice49.
KLOTHO, PHOSPHATE AND FGF23
Animal models lacking klotho or FGF23 develop similar phenotypes with growth retardation, shortened life span, phosphate retention, osteopaenia and vascular and ectopic calcifications, among other alterations, revealing an unexpected link between phosphate and aging50,51. These similar phenotypes of klotho and FGF23 knockout mice have been explained by the discovery that FGF23 requires klotho to bind with high affinity to the FGF receptor21.
However, in CKD patients, it is the increase of FGF23 levels and not FGF23 level decrease that is associated with mortality, higher risk of cardiovascular events and vascular calcifications41-47. This discrepancy with the FGF23 knockout phenotype may be explained by the finding that mice lacking klotho show, similarly, high levels of FGF2352 (Table I). It was hypothesised that klotho deficiency creates a tissue resistance to FGF23 which is responsible for the increase of FGF23 levels52. Reduced renal expression of klotho has been demonstrated in CKD patients53 preceding FGF23 increase54. Chronic kidney disease may be considered a state of klotho deficiency with increase of FGF23 levels similar to what is observed in klotho-deficient mice. The rescue of klotho-deficient or FGF23-deficient mice has been performed by correcting the hyperphosphataemia or hypervitaminosis D with dietary or genetic interventions.
Table I
CKD shares some manifestations of Klotho knockout and FGF23 knockout Models
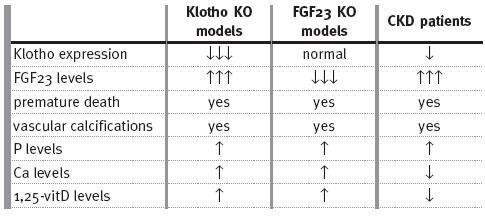
These mice do not develop vascular calcifications and show an increase in life span. All these interventions have in common the reduction of phosphate levels, with opposite effects on Ca and vitamin D levels, suggesting that phosphate is primarily responsible for these aging-like phenotypes54. Klotho deficiency may be the initial alteration for the development of phosphate retention and secondary hyperparathyroidism in CKD patients54.
PROPOSED ROLE OF KLOTHO AND FGF23 IN THE DEVELOPMENT OF SECONDARY HYPERPARATHYROIDISM IN CKD (Fig. 2)
Phosphate load in the diet transitorily stimulates FGF2323,25 with a consequent and adequate phosphaturic action25,26, contributing to the maintenance of normal phosphate blood levels. In CKD patients, with reduction of renal mass, there is a decrease in the renal expression of klotho53, contributing to the increased levels of FGF2352. In initial CKD stages, this increase of FGF23 levels maintains an efficient phosphate excretion and is associated with an increase of fractional excretion of phosphate and with normal phosphate levels. At the same time FGF23 decreases 1,25(OH)2D325,26 by decreasing its synthesis and increasing its catabolism. The decrease of 1,25(OH)2D3 is a stimulus for the increase in the synthesis of PTH7.
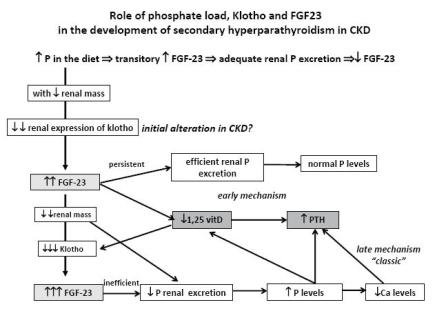
Role of phosphate load, klotho and FGF23 in the development of secondary hyperparathyroidism in CKD.
As the kidney insufficiency progresses there is a greater decrease of renal klotho expression to which 1,25(OH)2D3 also deficiency contributes. This decrease in klotho expression contributes to a further increase in FGF23 levels. At this stage, with important reduction of renal mass, FGF23 increase is no longer efficient, and reduced phosphaturia is responsible for the appearance of hyperphosphataemia.
In this proposed pathway for the development of secondary hyperparathyroidism two main mechanisms are recognised: an early mechanism resulting from the increase of FGF23, associated with low 1,25(OH)2D3 levels32 and a late mechanism, corresponding to the classic trade-off hypothesis derived from hyperphosphataemia. The decrease in the renal expression of klotho precedes the increase of FGF23 levels54 and may be the initial alteration in CKD patients responsible for the development of secondary hyperparathyroidism54.
CONCLUSIONS
FGF23 is a regulator of phosphate and vitamin D metabolism. FGF23 levels are increased in early CKD stages. Increase of FGF23 seems to be a novel mechanism for the early decline of 1,25(OH)2D3 levels observed in CKD patients. Phosphate is one of the recogniaed stimuli for FGF23 secretion. In this new updated model for secondary hyperparathyroidism, FGF23 may act as a link between phosphate load and low vitamin D levels [Figs 1 and 2]. The discovery of klotho and FGF23 actions has given back to phosphate a primordial role in the development of secondary hyperparathyroidism.
It is possible that in CKD stages3 and 4 an early therapeutic intervention on phosphate with a low phosphate diet and/or phosphate binders, even in the presence of normophosphataemia36, might retard the development of secondary hyperparathyroidism.
References
1. Silver J, Moallem E, Killav R, et al. New insights into the regulation of parathyroid hormone synthesis and secretion in chronic renal failure. Nephrol Dial Transplant 1996;11 (Suppl 3):2-5 [ Links ]
2. Haussler MR, Norman AW. Chromosomal receptor for a vitamin D metabolite. Proc Natl Acad Sci USA 1969;62:155-62 [ Links ]
3. Brown E, Gambia G, Riccardi D, et al. Cloning and characterization of an extracellular calcium sensing receptor from bovine parasthyroid. Nature 1993;366:575-580 [ Links ]
4. Kilav R, Silver J, Naveh-Many T. Parathyroid hormone gene expression in hypophosphatemic rats. J Clin Invest 1995;96:327-33 [ Links ]
5. Hernandez A, Concepcion MT, Rodriguez M, Salido E, Torres A. High phosphorus diet increases prepro PTHm RNA independent of calcium and calcitriol in normal rats. Kidney Int 1996;50:1872-8 [ Links ]
6. Kates DM, Sherrard DJ, Andress DL. Evidence that serum phosphate is independently associated with serum PTH in patients with chronic renal failure. Am J Kidney Dis 1997;30:809-13 [ Links ]
7. Silver J, Naveh-Many T, Mayer H, et al. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J Clin Invest 1986;78:1296-301 [ Links ]
8. Moallem E, Silver J, Kilav R, et al. RNA protein binding and post-transcriptional regulation of PTH gene expression by calcium and phosphate. J Biol Chem 1998; 273:5253-9 [ Links ]
9. Navey-Many T, Rahamimov R, Livni N, et al. Parathyroid cell proliferation in normal and chronic renal failure rats. The effects of calcium, phosphate and vitamin D. J Clin Invest 1995;96:1786-93 [ Links ]
10. Bricker NS: On the pathogenesis of the uremic state. An exposition of the trade-off hypothesis. N Engl J Med 1972;286:1093-9 [ Links ]
11. Slatopolsky E, Delmez JA: Pathogenesis of secondary hyperparathyroidism. Nephrol Dial Transplant 1996;11 (Suppl 3):130-5 [ Links ]
12. Levin A, Bakris GL, Molitch M. et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int 2007;71:31-8 [ Links ]
13. Kestenbaum B, Sampson JN, Rudser KD, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 2005;16520-8 [ Links ]
14. Llach F, Yudd M. Pathogenic, clinical, and therapeutic aspects of secondary hyperparathyroidism in chronic renal failure. Am J Kidney Dis 1998;32 2 Suppl 2):S3-S12 [ Links ]
15. Llach F. Secondary hyperparathyroidism in renal failure: the trade-off hypothesis revisited. Am J Kidney Dis 1995;25:663-79 [ Links ]
16. Llach F, Yudd M. Pathogenic, clinical, and therapeutic aspects of secondary hyperparathyroidism in chronic renal failure. Am J Kidney Dis 1998;32 (2 Suppl 2):S3-S12 [ Links ]
17. Kaye M. Hypocalcemia after an acute phosphate load is secondary to reduced calcium efflux from bone: studies in patients with minimal renal function and varying parathyroid activity. J Am Soc Nephrol 1995;6:273-80 [ Links ]
18. Isakova T, Gutierrez O, Shah A, et al. Postprandial mineral metabolism and secondary hyperparathyroidism in early CKD. J Am Soc Nephrol 2008;19:615-23 [ Links ]
19. Mohammadi M, Olsen S.K, Ibrahimi O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Ver 2005;16:107-37 [ Links ]
20. Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol 2001;2(3):reviews 3005 [ Links ]
21. Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 2006;281:6120-3 [ Links ]
22. Liu S, Zhou J, Tang W et al. Pathogenic role of FGF 23 in Hyp mice. Am J Physiol Endocrinol Metab 2006;291:E38-49 [ Links ]
23. Saito H, Maeda A, Ohtomo S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3and phosphorus in vivo. J Biol Chem 2005;280:2543-9 [ Links ]
24. Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 2006;17:1305-15 [ Links ]
25. Saito H, Kusano K, Kinosaki M, et al. Human fibroblast growth factor-23 mutants suppress Na-dependent phosphate co-transport activity and 1alpha,25- dihydroxyvitamin D3 production. J Biol Chem 2003;278:2206-11 [ Links ]
26. Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 2004; 19:429-35 [ Links ]
27. Kuro-o M. Klotho in chronic kidney disease – Whats new? Nephrol Dial Transplant 2009;24:1705-8 [ Links ]
28. Larsson T, Nisbeth U, Ljunggren O, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 2003;64:2272-9 [ Links ]
29. Gutierrez O, Isakova T, Rhee E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 2005;16:2205-15 [ Links ]
30. Evenepoel P, Meijers B, Viaene L, et al. Fibroblast growth factor-23 in early chronic kidney disease: additional supporting favor of a phosphate-centric paradigm for the pathogenesis of secondary hyperparathyroidism. Clin J Am Soc Nephrol 2010;5:1268-76 [ Links ]
31. Wolf M. Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol 2010;21:1427-35 [ Links ]
32. Gutiérrez OM. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: updating the trade-off hypothesis. Clin J Am Soc Nephrol 2010:5:1710-6 [ Links ]
33. Ito N, Fukumoto S, Takeuchi Y, et al. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab 2007; 25:419-22 [ Links ]
34 Nishida Y, Taketani Y, Yamanaka-Okumura H, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int 2006;70:2141-7 [ Links ]
35. Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol 2007;18:1637-47 [ Links ]
36. Moe SM, Zidehsarai MP, Chambers MA, et al.Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011;6:257-64 [ Links ]
37.Portale AA, Booth BE, Halloran BP, et al. Effect of dietary phosphorus on circulating concentrations of 1,25-dihydroxyvitamin D and immunoreactive parathyroid hormone in children with moderate renal insufficiency. J Clin Invest 1984; 73:1580-9 [ Links ]
38. Llach F, Massry SG. On the mechanism of secondary hyperparathyroidism in moderate renal insufficiency. J Clin Endocrinol Metab 1985;61:601-6 [ Links ]
39. Oliveira RB, Cancela AL, Graciolli FG, et al. Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin J Am Soc Nephrol 2010;5:286-91 [ Links ]
40. Isakova T, Gutiérrez OM, Smith K, et al. Pilot study of dietary phosphorus restriction and phosphorus binders to target fibroblast growth factor 23 in patients with chronic kidney disease. Nephrol Dial Transplant 2011;26:584-91 [ Links ]
41. Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008;359:584-92 [ Links ]
42. Jean G, Terrat JC, Vanel T, et al. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long hemodialysis patients. Nephrol Dial Transplant 2009;24:2792-6 [ Links ]
43. Parker BD, Schurgers LJ, Brandenburg VM, et al. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med 2010;18;152:640-8 [ Links ]
44.Seiler S, Reichart B, Roth D, et al. FGF-23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol Dial Transplant 2010 25:3983-9 [ Links ]
45.Gutierrez OM, Januzzi JL, Isakova T, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009;119:2545-52 [ Links ]
46. Jean G, Bresson E, Terrat JC, et al. Peripheral vascular calcification in long-hemodialysis patients: associated factors and survival consequences. Nephrol Dial Transplant 2009;24:948-55 [ Links ]
47. Fliser D, Kollerits B, Neyer U, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 2007;18:2600-8 [ Links ]
48. Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 2011:22:124-36 [ Links ]
49. Faul C,AmaralAP,Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011;121:4393-408 [ Links ]
50. Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997;390:45-51 [ Links ]
51. Shimada T, KakitaniM, Yamazaki Y, et al . Targeted ablation of FGF23 demonstrates as essential physiological role of FGF23 in phosphate and vitamin D metabolism. J ClinInvest 2004;113:561-8 [ Links ]
52.Urakawa I Yamazaki Y, Shimada T, et al. Klothoconverts canonical FGF receptor into a specific receptor for FGF23. Nature 2006;444:770-4 [ Links ]
53. Koh N, Fujimori T, Nishiguchi S et al. Severely reduced production of klothoin human chronic renal failure kidney. BiochemBiophys Res Commun2001;280:1015-20 [ Links ]
54. Kuro-o M. Phosphate and Klotho. Kidney Int2011;79(Suppl121):S20-S23 [ Links ]
Dra. Teresa Adragão
Nephrology Department, Hospital de Santa Cruz
Carnaxide, Portugal
Email:tadragao@netcabo.pt
Conflict of interest statement. None declared
Received for publication: 26/10/2011
Accepted in revised form: 28/02/2012

