










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.26 no.1 Lisboa jan. 2012
Multicystic dysplastic kidney: a review of eleven years (2000 – 2010)
Helena Rios, Raquel Santos, Clara Gomes, António Jorge Correia
Paediatric Nephrology
Centro Hospitalar de Coimbra. Coimbra, Portugal.
ABSTRACT
Introduction. Multicysticdysplastic kidney is the most common form of renal dysplasia. The natural history of multicystic dysplastic kidney is involution of the affected kidney, which justifies conservative management. The aim of this study was to evaluate the clinical course of a group of children with multicystic dysplastic kidney and compare these results with a previous study conducted 1989 -2000 in the same clinic.
Patients and Methods. Retrospective analysis of the medical records of all children with multicystic dysplastic kidney referred to the paediatric nephrology unit of a tertiary paediatric hospital 2000-2010.
Results. Fifty-two children (54% female) with multicystic dysplastic kidney were studied. The mean age at the time of the first visit was nineteen months with a mean follow-up time of sixty-five months. Prenatal ultrasound showed renal abnormalities in 96% of the children, 80% of which were suggestive of multicystic dysplastic kidney. All children underwent renal ultrasound and renal scintigraphy, 48 (92%) had a voiding cystourethrography and two (4%) intravenous urography. Ten (19%) children had contralateral kidney anomalies without obstruction of urinary tract, with the most frequent being pelvicalyceal dilatation (5) and vesicoureteral reflux (5).
Nephrectomy was performed in five children (10%). Indications for nephrectomy were increased multicystic dysplastic kidney size in two children, ureterocele in two and bladder diverticulum in one. Of the 47 children (90%) with conservative treatment, 21 (45%) had total kidney involution (over a mean follow-up of three years).
Ten (19%) children had complications, eight (15%) of them had urinary tract infection, and two proteinuria.
Conclusions. As in the 1989-2000 study, prenatal ultrasound was the main form of multicystic dysplastic kidney diagnosis and contralateral kidney anomalies remain frequent (19% vs.=29%). There has been a significant reduction in the number of intravenous urographies (4% vs. 45%). Conservative treatment has been the first choice in the last few years, accompanied by a reduction in nephrectomies (10% vs. 37%). The lack of clinical problems and good evolution of children with multicystic dysplastic kidney, justifies conservative management. As these children have only one functioning kidney, an adequate monitoring with renal ultrasound and assessment of blood pressure and renal function is essential.
Key Words: Children; multicystic dysplastic kidney; nephrectomy; renal dysplasia.
INTRODUCTION
Multicystic dysplastic kidney (MCDK) is defined as the most severe form of renal dysplasia1,2 in which the kidney consists of multiple noncommunicating cysts separated by minimal and nonfunctional dysplastic parenchyma1,2,3.
It is strongly suspected that MCDK is the major reason for having only one visible kidney, a common abnormality in the general population5.
The prevalence of unilateral MCDK is 1:4300 births1, with left kidney and males more often affected1,2. Bilateral MCDK is rare; 1:10000 live births1 and fatal.
The pathogenesis is not well established. It can result from anomalies in the renal development, probably related to genetic anomalies, teratogens due to congenital viral infections or medications, or obstruction in uteroof the urinary tract4.
Often the contralateral kidney exhibits some anomalies (in the literature, the prevalence of those lanomaliesvary between 15.3% and 42%)7, the most prevalent being vesicoureteral reflux and obstruction of the ureteropelvic junction1-3. All of these may predispose to urinary tract infections with renal scarring, leading to progressive renal insufficiency4,6.
In 15-20% of MCDK1, extrarenal malformations can be identified. These include heart defects, oesophageal or intestinal atresia, myelomeningococele and VATER association (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheo-oesophageal fistula, oesophageal atresia, kidney and/or radial anomalies and limb defects )1,2.
In more than half of MCDK, the diagnosis is made by prenatal ultrasonography as large peripheral cysts without recognisable renal parenchyma2.
The affected kidney is nonfunctional1-3,5, so evaluation of the contralateral kidney is mandatory with serial renal ultrasonography. Voiding cystourethrography should not be performed as routine, unless initial renal ultrasound shows anomalies in the contra lateral upper urinary tract and/or kidney or the child develops a urinary tract infection (UTI)2,4-6.
The natural history of MCDK is involution of the affected kidney1-3,6. Sixty percent will regress or involute within three years8 which gives a good prognosis to this pathology. The incidence of complications such as hypertension, proteinuria, UTI, renal insufficiency and neoplasia (i.e.Wilms tumour) is low2,6. Recent studies refer that the risk for hypertension may be no greater than that of general paediatric population6. The risk of UTI (5% )6 appears to be associated with vesicoureteral reflux or other coexisting abnormalities rather than the MCDK8. Concerns for potential malignant degeneration are not supported by many of the more current reviews6.
For all these reasons a nonsurgical approach is recommended as first-line therapy. Nephrectomy of the MCDK is an option when there are problems of mass effect, hypertension and potential for malignancy5.
The aim of this study was to evaluate the clinical course of a group of children with MCDK followed at the paediatric nephrology unit of a tertiary paediatric hospital between 2000 and 2010 and compare these results with a previous study conducted between 1989 and 2000 at the same unit.
Patients and Methods
We performed a retrospective analysis of medical records of children with MCDK who started followup at our unit between January 2000 and December 2010.
The study included children with diagnosis of MCDK by renal ultrasonography, which confirmed kidney with multiple noncommunicating cysts and absence of renal function by radioisotope scan3,6.
The following variables were analysed: age, gender, personal and family history, how and when the diagnosis was made, results of imaging studies, associated anomalies, treatment and follow-up.
We also compared our results with a previous study conducted between 1989 and 2000 at the same unit.
Definitions:
Complete involution: disappearance of MCDK as shown by renal ultrasound. Age at which MCDK was undetectable was accepted as the time of complete involution3,7.
Partial involution: lreduction in size of the MCDK as shown by renal ultrasound3,7.
Compensatory hypertrophy of contralateral kidney: bipolar diameter greater than two standard deviations of the mean value of normal kidney according to age4,5,7.
Urinary tract infection: positive urine culture associated with fever or urinary symptoms3.
Hypertension: lsystolic and/or diastolic blood pressure greater than or equal to the 95 th lpercentile (adjusted for age, gender and height) on at least three separate occasions9.
Proteinuria: lprotein/creatinine ratio (mg/mmol) on a spot urine sample >20 in children over two years of age and >50 in infants and toddlers aged 6 to 24 months10.
RESULTS
From January 2000 to December 2010 (eleven years), 52 children with MCDK were referred to the paediatric nephrology unit, with a median of five cases per year.
The majority of patients were referred from maternity (63%) and from secondary hospitals (17%).
There was no clear prevalence in gender (54% females). The mean age of the patients at the time of the first consult was 19 months (17 days to 8 years), with a median of six months. Fifty-four percent of children were aged less than 12 months old. Mean time of follow-up was sixty-five months.
In six children (12%), MCDK was associated with other congenital abnormalities: three with congenital heart defects, two with VATER syndrome and two with chromosome anomalies (one child with Triple X syndrome (47XXX) and another with Klinefelter syndrome).
Family history of renal pathology was positive in 15 (29%) children; two children with MCDK and two with single kidney.
Prenatal ultrasound showed renal anomalies in 50 (96%) children, and in 41 (82%) of them it revealed MCDK (Fig. 1). MCDK was diagnosed after birth in two children, assessment due to a urinary tract infection (at three years and five months old) and an oesophageal atresia (on the first day of life).
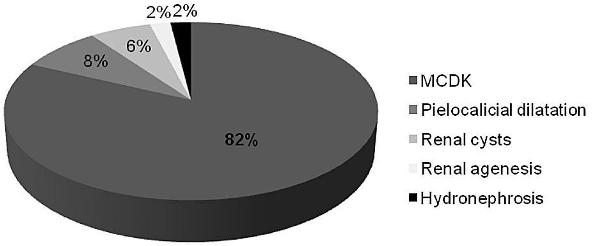
Figure 1
Prenatal diagnosis of renal anomalies (n=50).
All children had undergone renal ultrasound and renal scintigraphy (mercaptoacetyltriglycine (MAG3) or dimercaptosuccinic acid (DMSA)), 48 (92%) had a voiding cystourethrography and two (4%) an intravenous urography.
There was a predominance of left-side MCDK (58%). Imaging studies showed contralateral kidney abnormalities in ten (19%) children: five children with pelvicalyceal dilatation, five with vesicoureteral reflux, one with duplication of the collecting system and one with pelvic kidney. In one child a bladder diverticulum was also diagnosed.
Prophylactic antibiotics were prescribed in 38 (73%) children until investigation was completed (mean of nine months).
In 47 children (90%), the first-line treatment was conservative, and MCDK had spontaneous involution in 38 (81%) children (total involution in 21, 45%), over a mean follow-up of three years. Prenatal involution occurred in one child (Fig. 2).
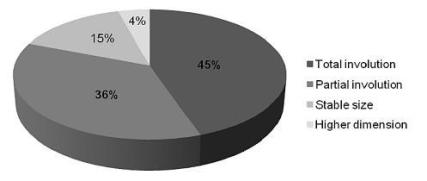
Figure 2
Evolution of renal sizes in children with MCDK with conservative treatment
Nephrectomywas performed in five children (10%) at a mean age of 23 months old. Indications for nephrectomy were increased MCDK size (2), ureterocele (2) and bladder diverticulum (1). In four children nephrectomy was performed in a conventional way and in the other by laparoscopy.
During the follow-up period, 10 (19%) children had complications. Eight (15%) had UTI (three of them with vesicoureteral reflux in contralateral kidney) and two had proteinuria. None of the patients subsequently developed hypertension, chronic renal disease or malignancy.
Compensatory hypertrophy of the contralateral kidney was noted in 46 (88%) children. Three of the six children without compensatory hypertrophy of the contralateral kidney had vesicoureteral reflux.
Five children (10%) were discharged and the rest remain in follow-up at the paediatric nephrology unit, with regular clinical and ultrasound evaluation.
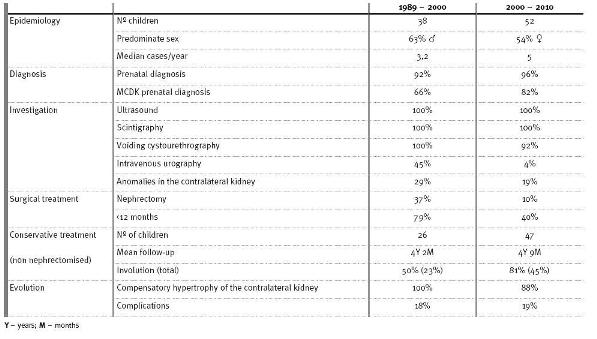
Table I compares the results of this study with the earlier one conducted in the same Paediatric Nephrology Unit between 1989 and 2000lang11.
Comparison of the children with MCDK referred to the Paediatric Nephrology Unit from 1989 to 2000 with those referred from 2000 to 2010
DISCUSSION
There was a slight predominance of females in our MCDK population, which was not consistent with the literature where males predominate3,6,7 . The diagnosis of MCDK was made in the majority of patients by prenatal ultrasound and was more frequent on the left side, which was consistent with other cohorts3,5-7.
As patients with MCDK have only one functional kidney, the anomalies in the contralateral kidney play a crucial role in the prognosis. Despite a similar prevalence of anomalies in the contralateral kidney (19%) compared to other studies (varying between 15.3% and 42% )7, presence of vesicoureteral reflux was lower (10% vs. 19%4,5 or 22%7).
Although in recent literature voiding cystourethrography only be recommended in the presence of renal ultrasound abnormities in the contralateral upper urinary tract and/or kidney or the child develops a UTI2,4-6, in our review a high percentage of children underwent voiding cystourethrography.
Compensatory hypertrophy of the contralateral kidney is generally expected in patients with MCDK: our result was 88%, in line with the literature (81%5 ; 89.8%7; 98.5%6).
As the MCDK often undergoes involution and complications such as malignant transformation and hypertension are rare, the option for a conservative follow-up is justifiable. The prevalence of nephrectomy in our study, 10%, was similar to other reports which vary between 5 and 19.8%3,5.
When comparing our study with another conducted at the same unit between 1989 and 200011(Table I), we emphasise the following aspects:
1. In recent years there has been a higher average of cases per year (5 vs. 3.2). The higher prevalence of MCDK could be explained by the advances in prenatal diagnosis3.
2. In both periods, prenatal ultrasound was the main form of MCDK diagnosis. However there has been a more accurate MCDK diagnosis in the last decade (82% vs. 66%), possibly due to advances in the diagnosis of fetal malformations by prenatal ultrasound.
3. Regarding the investigation, the main change was a significant reduction of intravenous urography (4% vs. 45%).
4. Contralateral kidney anomalies remain frequent (19% vs. l29%).
5. Conservative treatment has been the first choice in the last few years, confirmed by the reduction of nephrectomies (10% vs. 37%).
6. In the last 11 years the percentage of total involution in non nephrectomised patients has been higher (45% vs. 23%).
7. The incidence of complications was similar.
The lack of clinical complications justifies conservative management5, with adequate monitoring essential.
A child with MCDK must have periodic follow-up visits every 3-6 months during the first year and thenyearly to monitor for potential complications6 .This management includes growth-chart analysis, blood pressure assessment, urinary and blood tests and renal ultrasonography8. Ultrasound follow-up must assess for MCDK involution, contralateral kidney growth and screen for urinary tract abnormalities4,6.
However, based on the lack of significant complications and clinical symptoms, recommendations for ultrasound follow-up are quite variable4. Some authors suggest that it must be repeated at one and two years old and then every subsequent two or three years lang6; others prefer to do it every three-six months (first year), every six months (second year) and then annually8, and others suggest that, after the annual review at two years of age, the next clinical and ultrasound assessment could be carried out at five years and then at ten years 5. Renal function with measurement of serum creatinine should be evaluated at two, five and ten years of age if the contralateral kidney is normal4.
At any time of the follow-up, parents should beinformed that they should return sooner if the child has UTI, abdominal complaints, failure to thrive or aches5.
In the absence of renal insufficiency, proteinuria, hypertension or UTI with complete involution or a clear trend towards regression of MCDK and the contralateral kidney has reached compensatory hypertrophy, follow-up by a paediatric nephrologist with serial ultrasound would no longer be necessary3,6.
Renal ultrasound should be maintained at a general paediatric clinic, with screening for high blood pressure and proteinuria6.
In conclusion MCDK is a disease with a good prognosis and its natural history is progressive involution.
The lack of clinical problems justifies conservative management. As these children have only one functional kidney, an adequate monitoring with renal ultrasound and assessment of blood pressure and renal function is essential.
References
1 T. Prenatal sonographic diagnosis of cystic renal disease. UpToDate Version 19.2; Last update April 2011. "http://www.uptodate.com/contents/prenatal-sonographicdiagnosis-of-cystic-renal-disease?source=search_result&search=multicystic+dysplastic+kidney&selectedTitle=3%7E14". Accessed 5 August 2011
2 Niaudet P. Renal cystic diseases in children. UpToDate Version 19.2; Last update June 2011. "http://www.uptodate.com/contents/renal-cystic-diseases-in-children?source=search_result&search=multicystic+dysplastic+kidney&selectedTitle=1%7E14". Accessed 5 August 2011 [ Links ]
3 Mansoor O, Chandar J, Rodriguez MM, et al Long-term risk of chronic kidney disease in unilateral multicystic kidney. Pediatr Nephrol 2011;26:597-603 [ Links ]
4 Hains DS, Bates CM, Ingraham S. Management and etiology of the unilateral multicystic dysplastic kidney: a review. Pediatr Nephrol 2009;24:233-241 [ Links ]
5 Aslam M, Watson AR. Unilateral multicystic dysplasic kidney: long term outcomes. Arch Dis Child 2006;91:820-823 [ Links ]
6 Weinstein A, Goodman TR, Iragorri S. Simple multicystic kidney disease: end points for subspecialty follow-up. Pediatr Nephrol 2008 ;23:111-116 [ Links ]
7 Kiyak A, Yilmaz A, Turhan P, Sander S, Aydin G, Aydogan G. Unilateral multicystic dysplastic kidney: single-center experience. Pediatr Nephrol 2009;24:99-104 [ Links ]
8 Cambio AJ, Evans CP, Kurzrock EA. Non-surgical management of multicystic dysplastic kidney. BJU Int 2008 ;101:804-808 [ Links ]
9 Luma GB, Spiotta RT. Hypertension in children and adolescents. American Family Physician 2006 ;73:1559-1570 [ Links ]
10 Gagnadoux MF. Evaluation of proteinuria in children. UpToDate Version 19.2; Last update April 2010. "http://www.uptodate.com/contents/evaluation-of-proteinuria-in-children?source=search_result&search=proteinuria+definition+in+children&selectedTitle=1%7E150". Accessed 5 August 2011 [ Links ]
11 Gomes C, Timas R, Correia AJ. Clínica e evolução o rim multiquístico: que dilemas? Acta Pediatr Port 2001;6(32):363-366 [ Links ]
Helena Rios
Travessa do Cedro nº180, 2ºDt; 4535-246 Mozelos VFR
E-mail: helenarios@sapo.pt
Conflict of interest statement. None declared.
Received for publication:07/11/2011
Accepted in revised form:09/02/2012

