










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.27 no.3 Porto jun. 2013
ARTIGO DE REVISÃO
Síndrome de Wernicke-Korsakoff -revisão literária da sua base neuroanatómica
Wernicke-Korsakoff syndrome: a literary review on its neuroanatomical basis
André Silva1, André Enes1
1Faculdade de Medicina da universidade do Porto
RESUMO
O elevado consumo de álcool, não apenas por parte da população adulta mas também, e cada vez mais, por parte dos estratos mais jovens, apresenta-se como um problema de saúde pública preocupante. Uma das possíveis consequências do alcoolismo crónico é o desenvolvimento do síndrome de Wernicke-Korsakoff. O síndrome de Wernicke-Korsakoff é um grupo de sinais e sintomas neuropsiquiátricos induzido por uma deficiência nutricional de vitamina B1 (tiamina). Esta doença, sem um tratamento adequado, pode progredir para um estado de estupor, coma e até morte. Os autores esclarecem a sua fisiopatologia e manifestações e, no processo, foi realizada uma revisão literária.
Palavras-chave: Encefalopatia de Wernicke, Síndrome Korsakoff, alcoolismo, estado nutricional
ABSTRACT
High consumption of alcohol, not only by the adult population but also, and ever increasing, by younger strata, presents itself as a worrisome public health issue. One of the possible consequences of chronic alcohol consumption is the development of the Wernicke-Korsakoff syndrome. it is a group of neuropsychiatric signs and symptoms induced by a nutritional deficiency of vitamin B1 (thiamine). This disease, without proper treatment, can progress to a state of stupor, coma and even death. The authors shed a light on its pathophysiology and manifestations and, in the process, a literary review was undertaken.
Key-words: Wernicke encephalopathy, Korsakoff Syndrome, alcoholism, nutritional status
INTRODUÇÃO
A mal nutrição representa um problema de saúde pública relevante nos países em desenvolvimento, no entanto a sua expressão nos países desenvolvidos não é negligenciável. O défice nutricional pode ser, de facto, um factor precipitante de distúrbios neurológicos. O síndrome de Wernicke-Korsakoff foi inicialmente reconhecido como uma entidade clínica nos finais do século XIX graças aos contributos dos clínicos Carl Wernicke e Sergey Korsakoff.1 Refere-se a uma constelação de sinais e sintomas neuropsiquiátricos que resultam de uma deficiência nutricional em tiamina (vitamina B1).1
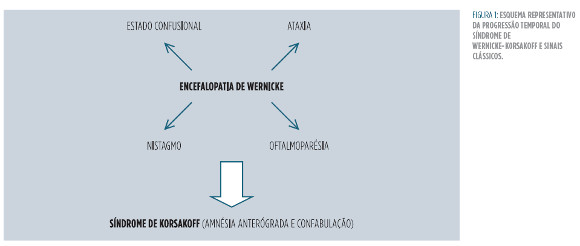
Consiste em duas fases distintas (Figura 1 ) de um mesmo processo patológico: inicialmente surge, mas nem sempre, a encefalopatia de Wernicke (fase aguda do síndrome) caracterizada pela tríade clínica clássica de estado confusional agudo (perturbação aguda e flutuante da atenção e do correto processamento dos estímulos originados do meio externo), oftalmoparésia (parésia de um ou mais músculos extra-oculares), e ataxia (perda da coordenação motora).1 O nistagmo (movimentos oculares involuntários e oscilatórios) também é característico desta fase, apesar de não constar na descrição clássica. Com a progressão do processo patológico, a encefalopatia pode progredir para um quadro crónico – síndrome de Korsakoff – marcado por uma amnésia anterógrada (incapacidade de formar novas memórias) e confabulação (produtos falsos da memória), à medida que os sinais da primeira subsidem.1 Se a identificação e abordagem terapêutica deste síndrome forem tardias poderão surgir estupor, coma e, eventualmente, a morte.
MÉTODOS
Artigos foram encontrados através dos motores de busca electrónicos ISI Web of Knowledge e pubmed. As referências pertinentes para os objectivos estabelecidos foram analisadas e incluídas na revisão da literatura. Foram considerados todos os artigos desde o ano de 1950 até ao tempo presente e a busca foi limitada a artigos escritos em inglês. Os termos usados na busca foram: ((Wernicke-Korsakoff Syndrome) Or (Wernicke Encephalopathy) Or (Korsakoff Syndrome)) and ((imaging) Or (neuroimaging) Or (anatomical) Or (pathology) Or (neuropathology) Or (biochemical)).
De um total de 824 artigos foram considerados relevantes 36 artigos que foram, ultimamente, usados nesta revisão. a leitura de alguns dos artigos encontrados suscitou a consulta de outras obras referenciadas nos primeiros que os autores deste artigo consideraram relevantes para a exploração do tema.
EPIDEMIOLOGIA
A tríade característica da fase aguda só é constatada em cerca de 16% dos doentes2, facto este que acrescenta dificuldades no diagnóstico do síndrome. de facto, este é realizada com sucesso numa pequena fração dos pacientes(20%).3 Por outro lado, os sinais e sintomas poderão ser confundidos com os efeitos da intoxicação alcoólica aguda, o que poderá explicar que a proporção de casos confirmados, em autópsia, de encefalopatia de Wernicke (0.8% – 2.8%) seja superior à estimada no diagnóstico clínico (0.04% – 0.13%).4,5 Esta constatação é merecedora de uma reflexão já que, segundo a Organização mundial da Saúde (OMS), Portugal é dos países onde se verifica um maior consumo de álcool per capita.40 Em boa verdade, os valores referidos acima foram obtidos, em grande parte, a partir de indivíduos alcoólicos, no entanto, é curioso que não existe uma correlação entre a prevalência da encefalopatia e o consumo de álcool per capita em vários países ocidentais.5 O síndrome apresenta uma mortalidade de 17%, sendo mais prevalente em homens do que em mulheres. Dos indivíduos com encefalopatia que sobrevivem (valor estimado em 80%) acabam por desenvolver o síndrome de Korsakoff.6,7 Esta conclusão é infeliz dado que a progressão da patologia pode ser travada com a administração de tiamina parenteral. Em seguida, encontram-se alguns factores que podem precipitar o surgimento deste síndrome.
ACHADOS BIOQUÍMICOS
O conceito de deficiência nutricional não pode ser apenas circunscrito à carência de um ou mais nutrientes essenciais na dieta, mas também abranger qualquer factor que condicione um aumento das necessidades do nutriente em questão.
A tiamina (vitamina B1) é uma das vitaminas essenciais do complexo B, possuindo um papel central no catabolismo de hidratos de carbono e formação de neurotransmissores.22 Apesar deste cofactor e respectivas enzimas estarem presentes em todas as células, as cardíacas e nervosas parecem particularmente sensíveis aos efeitos da deficiência de tiamina.30 a utilização desta vitamina depende da taxa metabólica do indivíduo, aumentando com uma maior necessidade energética.22 No tubo digestivo, este nutriente é absorvido activamente ao nível do duodeno, sendo posteriormente transportado através da barreira hemato-encefálica por processos passivos e activos.23 na sua forma biologicamente activa (tiamina pirofosfato), é uma coenzima essencial para várias enzimas do catabolismo da glicose-6-fosfato, tais como a transcetolase, a desidrogénase do piruvato e a desidrogénase do a-cetogluratato.23,28,30 A primeira enzima participa na via das pentoses-fosfato e da sua actividade catalítica resultam as moléculas ribose-5-fosfato e nicotinamida adenosina dinucleótido reduzida (NADPH). Ambas são fulcrais na síntese de vários outros compostos (ex: ácidos nucleicos e glutationa) e qualquer célula requer níveis óptimos destas enzimas. As outras duas enzimas catalizam reacções da glicólise e do ciclo de Krebs, respectivamente. Destas vias metabólicas resultam a formação de moléculas de adenosina trifosfato (ATP), essenciais no fornecimento de energia para o metabolismo celular. Níveis reduzidos das referidas enzimas conduzem a uma menor síntese energética e morte celular.30 O ser humano possui cerca de 30-50mg em reservas de tiamina, que se estima poder terminar em 2 a 3 semanas.28 por outro lado, as necessidades de tiamina aumentam com o abuso do álcool e aumento da ingestão de hidratos de carbono, dado que o primeiro é catabolizado de forma análoga a um glícido. Assim, entende-se que a combinação de uma dieta desequilibrada, assim como uma absorção gastrointestinal, armazenamento hepático e utilização cerebral de tiamina comprometidas, como se verifica no alcoolismo crónico, potencie o desenvolvimento do síndrome.
Num contexto de défice de tiamina, os sistemas bioquímicos dependentes desta e, por extensão, o metabolismo energético, estão comprometidos.29 Os mecanismos conducentes ao desenvolvimento de lesões no sistema nervoso ainda não estão inteiramente clarificados, no entanto, tem sido proposto que os mecanismos celulares da encefalopatia de Wernicke envolvem principalmente a perda localizada da barreira hemato-encefálica28,30 e a incapacidade da regiões encefálicas metabolicamente mais activas (ou seja, com um turnover de tiamina mais elevado) de manterem os gradientes osmóticos e, por conseguinte, a homeostasia de fluído intersticial.28,30 Em peças de autópsia, verifica-se nestas regiões (explicitadas na próxima secção): hemorragias petequiais, gliose, hipertrofia do endotélio e morte celular. Outros mecanismos, como o aumento da produção de espécies reactivas de oxigénio e excitotoxicidade, também já foram implicados com a deficiência em tiamina.28,30
NEUROPATOLOGIA
Em termos macroscópicos, as lesões mais frequentes encontram-se no núcleo talâmico dorso medial (bilateralmente), nos corpos mamilares, na substância cinzenta peri-aquedutal e no verme superior do cerebelo.7,24 Observa-se também um alargamento da fissura inter-hemisférica dos lobos frontais.25 No entanto, também podem existir outros locais afectados como o tegmento pôntico, a formação reticular do mesencéfalo e o córtex cerebral.7
Uma análise histopatológica releva a existência de variações, conforme se trate de uma lesão aguda ou de uma lesão crónica. no primeiro caso, observa-se uma distribuição simétrica de pequenas hemorragias no tronco cerebral e tálamo, sem infiltração de macrófagos e sem proliferação capilar relevante.26 por outro lado, numa lesão crónica observam-se edema citotóxico com balonização astrocitária, diminuição das fibras mielinizadas, activação da microglia, astrogliose reactiva e proliferação e engorgitamento microvascular, apesar de a perda de neurónios ser pouco relevante.7,27
No entanto, torna-se necessário realçar que as alterações macro e microscópicas dependem do estado e da severidade da encefalopatia de Wernicke.27
FISIOPATOLOGIA
As lesões da encefalopatia de Wernicke são tipicamente simétricas e localizam-se na proximidade do plano mediano.31,35 Relativamente às possíveis manisfestações do síndrome temos:
Amnésia anterógrada e confabulação (síndrome de korsakoff)
Os corpos mamilares são eminências hemisféricas localizadas no diencéfalo, anteriormente à substância perfurada posterior, cada um encapsulando um núcleo envolvido por fibras derivadas do fornix. Os corpos mamilares estão relacionadas como circuito de Papez (Figura 2 ).
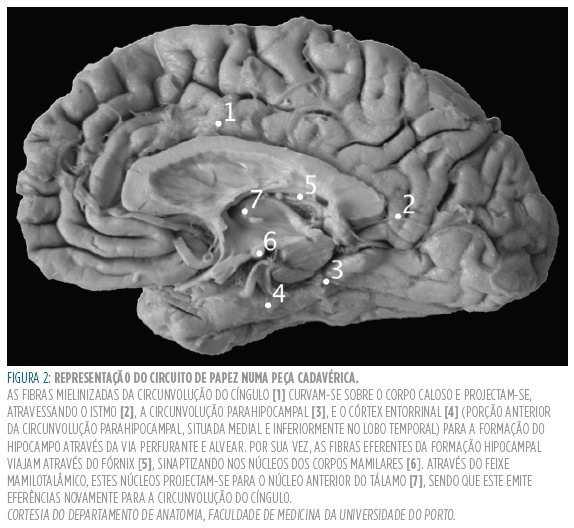
Uma lesão nos corpos mamilares terá como consequência a interrupção deste circuito. Como este circuito serve como substrato anatómico para a formação de novas memórias, uma interrupção deste causará uma amnésia anterógrada (incapacidade de formar novas memórias). Para além disto, os danos nos corpos mamilares poderão contribuir para o estado confusional que estes doentes apresentam no início da doença.
Alteração do estado de consciência, coma e síndrome confusional
A porção medial de ambos tálamos (grupos nucleares mediais e da linha média) apresentam ligação com o sistema reticular activador ascendente, cuja origem se localiza na transição pontomesencefálica do tegmento do tronco cerebral. Este último, é responsável pela regulação do estado de vigília. Assim, as lesões talámicas mediais bilaterais típicas desta encefalopatia, resultam em alterações no estado de consciência e atenção que podem apresentar-se desde a sonolência até ao coma e/ou estado confusional agudo.
Para além desta observação, a lesão neste local, assim como no hipotálamo e substância periaqueductal, que formam uma parte integrante do sistema de controlo central do sistema nervoso autónomo, pode precipitar um quadro de disautonomia com hipotermia, taquicardia e labilidade tensional arterial que surgem nalguns dos casos.28
Ataxia axial e de Marcha
O cerebelo recebe informação proprioceptiva inconsciente dos músculos do tronco veiculada pelos tratos espinhocerebelosos e projeta as suas eferências tanto para o núcleo vestibular e formação reticular através do núcleo fastigial como também, de forma directa, para os núcleos vestibulares. Os tratos reticuloespinal e vestibuloespinal que surgem dos núcleos referidos, por sua vez, têm a capacidade de influenciar os neurónios motores espinais. Sendo assim, faz sentido inferir que este tem um papel principal na regulação da postura e dos movimentos estereotipados que são programados pelo tronco cerebral e medula espinal.
A deficiência prolongada de tiamina provoca degeneração do córtex cerebeloso que começa nas porções mais anteriores do cerebelo e progride para regiões mais posteriores.32 O verme cerebeloso superior é, portanto, uma das estruturas commumente afectadas, sendo que os membros inferiores estão representados mais anteriormente no córtex cerebeloso. por conseguinte, as lesões no verme visíveis neste doentes poderão justificar a presença destes sinais. Ainda mais, as ligações do sistema vestibular também se encontram afectadas (vide infra) pelo que podem também contribuir para o desequilíbrio observado nestes casos.
Oftalmoparésia e nistagmo
O núcleo do nervo oculomotor localiza-se próximo do plano mediano, ventralmente na substância cinzenta periaquedutal. Consiste numa série de colunas longitudinais (subnúcleos), cada um deles responsável pela enervação de um músculo específico (recto superior, recto inferior e recto medial), enervando também o elevador da pálpebra superior e o oblíquo inferior. a oftalmoparésia resultante da lesão destes núcleos é variada e determinada pelos subnúcleos atingidos. O núcleo oculomotor acessório (Edinger-Westphal) contribui com fibras parassimpáticas para os músculos esfíncter da pupila e o músculo ciliar. A parésia do primeiro gera uma reposta fotomotora diminuída e lenta e a do ultimo um defeito da acomodação. Contudo, estes doentes apresentam mais frequentemente lesões periaqueductais que atingem de modo preferencial as áreas pré-tectais.28,33 Estas são áreas que estabelecem importantes ligações no circuito do reflexo fotomotor direto e consensual. Dado que o circuito do reflexo da acomodação se encontrasse parado do fotomotor, pode surgir no mesmo doente uma dissociação da resposta à luz (ausente) e na acomodação-convergência (preservado).28,33
O núcleo do nervo abducente localiza-se na parte caudal da protuberância, próximo do pavimento do quarto ventrículo, sendo que o colículo facial marca a sua localização. Este enerva somente o recto lateral. Na porção mais mediana de cada hemiprotuberância existe uma área designada por formação reticular paramediana pôntica que contem os programas motores para a execução dos movimentos conjugados horizontais ipsilaterais. Dado que o tegmento pôntico e os núcleos abducentes são os mais afectados, a alteração da oculomotricidade mais frequentes são o estrabismo medial bilateral em consequência da paralisia de ambos os rectos laterais e também da parésia do olhar conjugado horizontal.
O complexo nuclear vestibular localiza-se junto do pavimento do 4º ventrículo e recebe fibras aferentes do nervo vestibular provenientes do utrículo e do sáculo, assim como dos canais semicirculares. As fibras das duas primeiras estruturas conduzem informação sobre movimentos lineares da cabeça enquanto que os últimos informam sobre movimentos angulares da cabeça. Este complexo projecta eferências para vários locais no encéfalo, comunica nomeadamente com os núcleos dos nervos oculomotor, troclear e abducente (responsáveis pela totalidade dos movimentos do globo ocular) através do fascículo longitudinal medial. As ligações referidas permitem que os movimentos da cabeça e dos olhos sejam coordenados por forma a que a fixação visual num objecto possa ser mantida.
As lesões do complexo nuclear vestibular, cerebelo, tegmento pôntico e o fascículo longitudinal medial justificam o surgimento de nistagmo.28,33 Este pode apresentar fenomologia variada mas cuja base fisiopatológica se explica pela perda do reflexo, designado por vestíbulo-ocular, cuja função é de coordenação entre os movimentos oculares e da cabeça.
DIAGNÓSTICO
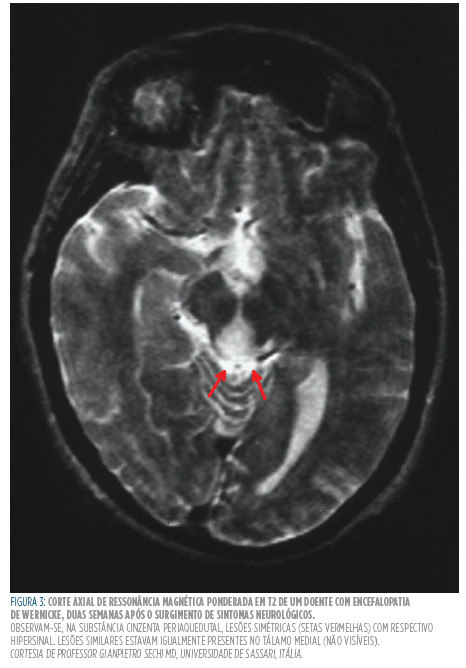
Como já for referido anteriormente, o subdiagnóstico desta patologia pode ser parcialmente explicado tanto pela variabilidade de apresentações clínicas como também devido à baixa especificidade dos sinais neurológicos.33,34 na fase aguda de deficiência em tiamina, os sintomas e sinais apresentados são vagos, podendo existir envolvimento neurológico (anormalidades oculares, estado mental alterado, estupor, crises epilépticas, halucinações) e cardiovasculares (hipotensão etaquicardia, insuficiência cardíaca). Já numa fase crónica, estes são mais específicos existindo confabulações e uma perda marcada da memória anterógrada comparativamente às restantes capacidades cognitivas.28 O seu diagnóstico é essencialmente clínico uma vez que não existem exames de rotina específicos que possibilitam a sua despistagem.28,34 Sendo assim, o clínico deve suspeitar a presença do síndrome em indivíduos que se apresentem malnutridos ou condições que amplifiquem a taxa metabólica ou interfiram com os processos de ingestão, digestão e absorção dos alimentos.35 O diagnóstico pode ser feito ao determinar a concentração sérica de tiamina, dos níveis de actividade da transcetolase nos eritrócitos, mas dada sua inexistência na maior parte dos hospitais e baixa sensibilidade e especificidade, a ressonância magnética cerebral pode ser útil para confirmar a suspeita clínica (Figura 2 ).28,38 A ressonância magnética (RM), com a sua elevada sensibilidade à presença de água no espaço intersticial, tornou possível uma melhor visualização dos sinais radiológicos da neuropatologia subjacente. Em algumas sequências, as lesões edematosas são reveladas ao observador como hipersinal, já que estas possuem um elevado teor em água. Enquanto que a sensibilidade da RM na detecção da encefalopatia é de apenas 53%, a sua especificidade é de 93%, o que permite que os seus achados sejam sugestivos da presença da patologia.35 Apesar da importância da ressonância magnética no diagnóstico do síndrome, os locais da lesões e as características do sinal encontrados não são patognomónicos da encefalopatia de Wernicke, por conseguinte, outras causas de encefalopatia aguda têm de ser consideradas, tais como o síndrome de Miller-Fisher, linfoma cerebral primário, doença de Behçet e ventrículoencefalite, entre outras. Outras condições como o infarto talâmico bilateral (síndrome do topo da basilar), lesão hipocampal após paragem cardiorespiratória e tumores do terceiro ventrículo devem ser englobados no diagnóstico diferencial devido à sobreposição neuroanatómica das lesões resultantes com as da encefalopatia de Wernicke.36,37
TRATAMENTO
A administração imediata de tiamina parenteral intravenosa ou intramuscular (500mg, 2 a 3 vezes por dia, durante 3 dias), seguida de uma suplementação oral diária – quando este iniciar a alimentação oral39 – pode permitir uma reversão gradual dos sinais da encefalopatia de Wernicke. No entanto, uma deficiência em tiamina demasiado prolongada leva a que esta terapêutica se revele ineficaz na resolução dos sinais da síndrome de Korsakoff, podendo até levar à morte do doente.39 Não é recomendada a administração de soro glicosado sem infusão de tiamina concomitante, já que pode precipitar um uma encefalopatia de Wernicke.39 A mensagem mais importante é de que não se deve esperar pela confirmação do diagnóstica para dar início ao tratamento.
CONCLUSÃO
Como já foi discutido, Portugal consta na lista de países onde se regista um maior consumo de álcool.40 Apesar deste não estar necessariamente implicado no surgimento do síndrome, o seu papel na desregulação da homeostasia da tiamina não são passíveis de serem ignorados.
Por outro lado, apesar de o síndrome encontrar-se mais associado ao alcoolismo crónico, pode surgir noutros contextos (vide supra) pelo que, nestas situações, deve ser considerado no diagnóstico diferencial do clínico.
AGRADECIMENTOS
Gostaríamos de expressar a nossa gratidão ao professor Gianpietro Sechi da Universidade de Sassari por nos ter gentilmente cedido a imagem de ressonância magnética ponderada em T2 (Figura 3) e, de igual modo, o serviço de Anatomia da Faculdade de Medicina da Universidade do Porto ao possibilitar a publicação da imagem da peça cadavérica (Figura 2).
REFERÊNCIAS
1. Victor M, Adams Ra, Collins GH. The Wernicke Korsakoff syndrome and related disorders due to alcoholism and malnutrition. 2nd Ed. Philadelphia: fa davis co; 1989 [ Links ]
2. Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J neurol neurosurg psychiatry 1986; 49: 341–45. [ Links ]
3. Kopelman md, Thomson ad, Guerrini i, marshall EJ. The Korsakoff syndrome: clinical aspects, psychology and treatment. alcohol alcohol. 2009; 44: 148–54. [ Links ]
4. Vasconcelos Mm, Silva Kp, Vidal G, Silva Af, Domingues Rc, Berditchevsky Cr. Early diagnosis of pediatric Wernickes encephalopathy. pediatr neurol 1999; 20: 289–94. [ Links ]
5. Harper C, Fornes P, Duyckaerts C, Lecomte S, Hauw JJ. An international perspective on the prevalence of the Wernicke-Korsakoff syndrome. metab Brain dis. 1995;10(1):17-24. [ Links ]
6. Torvik A. Wernickes encephalopathy-prevalence and clinical spectrum. Alcohol alcohol Suppl 1991; 1:381. [ Links ]
7. Victor M, Adams Rd, Collins GH. The Wernicke-Korsakoff syndrome: a clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp neurol Ser 1971; 1–206. [ Links ]
8. Heap Lc, Pratt OE, Ward RJ, et al. individual susceptibility to Wernicke-Korsakoff syndrome and alcoholism-induced cognitive deficit: impaired thiamine utilization found in alcoholics and alcohol abusers. Psychiatr Genet 2002; 12:217. [ Links ]
9. Chaves lc, faintuch J, Kahwage S, alencar fde a. A cluster of polyneuropathy and Wernicke-Korsakoff syndrome in a bariatric unit. Obes Surg 2002; 12:328. [ Links ]
10. Peltier G, Hermreck as, moffat re, et al. Complications following gastric bypass procedures for morbid obesity. Surgery 1979; 86:648. [ Links ]
11. Singh S, Kumar a. Wernicke encephalopathy after obesity surgery: a systematic review. Neurology 2007; 68:807. [ Links ]
12. Shorey J, Bhardway n, loscalzo J. Acute Wernickes encephalopathy after intravenous infusion of high-dose nitroglycerin. Ann intern med 1984; 101: 500 [ Links ]
13. Kwee il, nakada t. Wernickes encephalopathy induced by tolazamide. N Engl J med 1983; 309: 599–600. [ Links ]
14. Mclean J, manchip S. Wernickes encephalopathy induced by magnesium depletion. Lancet 1999; 353:1768. [ Links ]
15. Hung Sc, Hung SH, tarng dc, et al. Thiamine deficiency and unexplained encephalopathy in hemodialysis and peritoneal dialysis patients. Am J Kidney dis 2001; 38:941. [ Links ]
16. Patchell ra, fellows Ha, Humphries ll. Neurologic complications of anorexia nervosa. Acta neurol Scand. 1994;89:111–116. [ Links ]
17. Engel pa, Grunnet m, Jacobs B. Wernicke-Korsakoff syndrome complicating t-cell lymphoma: unusual or unrecognized? South med J 1991; 84:253. [ Links ]
18. Pittella JE, de castro lp. Wernickes encephalopathy manifestedas Korsakoffs syndrome in a patient with promyelocytic leukemia.South med J 1990; 83:570. [ Links ]
19 . Soffer d, Zirkin H, alkan m, Berginer Vm. Wernickes encephalopathy in acquired immune deficiency syndrome (aids): a case report. Clin neuropathol 1989; 8:192. [ Links ]
20. Gárdián G, Vörös E, Járdánházy t, et al. Wernickes encephalopathy induced by hyperemesis gravidarum. Acta neurol Scand 1999; 99:196. [ Links ]
21. Spruill Sc, Kuller Ja. Hyperemesis gravidarum complicated by Wernickes encephalopathy. Obstet Gynecol 2002; 99:875. [ Links ]
22. Tanphaichitr, V. Thiamin. In: Shils me, Olson Ja, Shike m, ross ac, editors. Modern nutrition in Health and disease. 9th ed. Baltimore: Williams and Wilkins; 1999: p. 381–89. [ Links ]
23. Thomson ad, cook cch, touquet r, Henry Ja. The royal college of physicians report on alcohol: guidelines for managing Wernickes encephalopathy in the accident and emergency department. Alcohol alcohol Suppl 2002; 37: 513–21. [ Links ]
24. Cochrane Wa, Collins-Williams C, Donohue Wl. Superior hemorrhagic poliencephalitis (Wernickes disease) occurring in an infant – probably due to thiamine deficiency from use of a soya bean product. Pediatrics 1961; 11: 771-77 [ Links ]
25. Lishman, W. A. Alcohol and the brain. Br. J. Psychiatr., 1990; 156: 5-17. [ Links ]
26. Vortmeyer Ao, Hagel C, Laas R. Haemorrhagic thiamine deficient encephalopathy following prolonged parenteral nutrition. J neurol neurosurg psychiatry 1992; 55: 826-29. [ Links ]
27. Harper c, Butterworth r. Nutritional and metabolic disorders. In: Vinken pj, Bruyn GW, eds. Handbook of clinical neurology, vol 28. Amsterdam: Elsevier, 1997: 243-70. [ Links ]
28. Sechi G, Serra A. Wernickes encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet neurol 2007. 6: 442–455 [ Links ]
29. Schenker S, Henderson Gi, Hoyumpa am jr, mc candless dw. Hepatic and Wernickes encephalopathies: current concepts of pathogenesis. Am J clin nutr 1980; 33: 2719–26. [ Links ]
30. Martin pr, Singleton ck, Hiller-Sturmhöfel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol res Health 2003; 27:134. [ Links ]
31. Malamud, n, Skillicorn, Sa. Relationship between the Wernicke and the Korsakoff syndrome: a clinicopathologic study of seventy cases. Arch neurol psychiat 1956; 76:586. [ Links ]
32. Mulholland pj. Susceptibility of the cerebellum to thiamine deficiency. Cerebellum 2006;5:55–63. [ Links ]
33. Zuccoli G, pipitone n. Neuroimaging findings in acute Wernickes encephalopathy: review of the literature. Ajr am J roentgenol 2009; 192:501. [ Links ]
34.Thomson ad, marshall EJ.The natural history and pathophysiology of wernickes encephalopathy and Korsakoffs psychosis. Alcohol alcohol 2006; 41: 151–58. [ Links ]
35. Sullivan EV, Pfefferbaum A. Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol alcohol 2009;44:155-65. [ Links ]
36. Yoneoka Y, Takeda N, Inoue A, et al. Acute Korsakoff syndrome following mammillothalamic tract infarction. Ajnr am J neuroradiol 2004; 25:964. [ Links ]
37. Renou P, Ducreux D, Batouche F, Denier C. Pure and acute Korsakoff syndrome due to a bilateral anterior fornix infarction: a diffusion tensor tractography study. Arch neurol 2008; 65:1252. [ Links ]
38. Caine D, Halliday Gm, Kril JJ, Harper cg. Operational criteria for the classification of chronic alcoholics: identification of Wernickes encephalopathy. J neurol neurosurg psychiatry 1997; 62:51. [ Links ]
39. Cook CC, Hallwood Pm, Thomson Ad. B Vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol alcohol 1998; 33:317. [ Links ]
40. Alcohol per capita consumption, patterns of drinking and abstention worldwide after 1995. Appendix 2. European addiction research, 2001, 7(3):155–157. [ Links ]
André Silva
Faculdade de Medicina da Universidade do Porto
Al. Prof. Hernâni Monteiro 4200-319 Porto
Email: mimed10263@med.up.pt

