










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArquivos de Medicina
versão On-line ISSN 2183-2447
Arq Med vol.26 no.6 Porto dez. 2012
ARTIGO DE REVISÃO
Síndrome MEN Tipo 2
MEN type 2 syndrome
Pedro Rodrigues1,2, José Luís Castedo1
1 Serviço de Endocrinologia, Diabetes e Metabolismo do Centro Hospitalar de São João E.P.E., Porto
2 Faculdade de Medicina da Universidade do Porto
RESUMO
A neoplasia endócrina múltipla tipo 2 é uma síndrome autossómica dominante causada por mutações germinativas do proto-oncogene RET. Tem uma penetrância elevada de carcinoma medular da tiróide e pode estar associada a feocromocitoma e hiperparatiroidismo. As principais formas clínicas descritas são: MEN 2A, MEN 2B e carcinoma medular da tiróide familiar. Cada variante resulta de mutações diferentes, havendo uma boa correlação genótipo-fenótipo em relação à idade de início e agressividade do carcinoma medular da tiróide e à presença de outros tumores endócrinos. As mutações do RET foram classificadas em quatro categorias de risco, com recomendações distintas a propósito da idade da tiroidectomia profilática. A MEN tipo 2 é um exemplo da importância do estudo genético no diagnóstico e tratamento precoces dos portadores.
Palavras-chave: Men 2; carcinoma medular da tiróide; mutações do gene ret; tiroidectomia
ABSTRACT
Multiple endocrine neoplasia type 2 is an autosomal dominant syndrome caused by germline mutations of RET proto-oncogene. it has a high penetrance of medullary thyroid carcinoma and may be associated with pheochromocytoma and hyperparathyroidism. MEN 2A, MEN 2B and familial medullary thyroid carcinoma are the main clinical forms described. Each variant comes from different mutations, showing a good genotype-phenotype correlation with regard to age of onset and aggressiveness of medullary thyroid carcinoma and presence of other endocrine tumours. RET mutations were classified into four risk categories with distinct recommendations on timing of prophylactic thyroidectomy. MEN type 2 provides a model for the importance of genetic testing in early diagnosis and treatment of mutation carriers.
Key-words: Men 2; medullary thyroid carcinoma; ret gene mutations; thyroidectomy
Introdução
As neoplasias endócrinas múltiplas (MEN, Multiple Endocrine Neoplasia) são um grupo de síndromes genéticas, com um padrão de transmissão autossómico dominante, que se caracterizam pelo aparecimento de tumores benignos ou malignos, envolvendo duas ou mais glândulas endócrinas. Existem dois tipos principais: a síndrome MEN tipo 1 e a síndrome MEN tipo 2.1
O tipo de mutação germinativa que está na origem destas síndromes é distinto. Enquanto na síndrome MEN tipo 1 há a ocorrência de mutações inactivadoras que envolvem a perda de genes de supressão tumoral, na síndrome MEN tipo 2 existem mutações activadoras que levam à formação de proto-oncogenes.2
A síndrome MEN tipo 1 caracteriza-se pela presença de tumores nas paratiróides, no pâncreas endócrino e na hipófise anterior. A síndrome MEN tipo 2 envolve o aparecimento de tumores na tiróide, na medula da suprarrenal e nas paratiróides. Apresenta-se uma revisão das manifestações clínicas, do diagnóstico e da abordagem terapêutica da síndrome MEN tipo 2.
Classificação
Até à data, a síndrome MEN tipo 2 foi identificada em 500 a1000 famílias anívelmundial.3 Temuma prevalência estimada de 2,5 por 100,000 na população geral e apresenta igual distribuição por género.4 Em termos geográficos, a maioria das famílias afectadas tem sido descrita na Europa, América do Norte e do sul e Austrália. Existem poucos casos registados na Ásia, excepto no Japão. No continente africano, a informação a propósito da incidência da síndrome é escassa.5
A síndrome MEN tipo 2 é classificada em dois subtipos principais: MEN2A e MEN2B (Tabela 1).
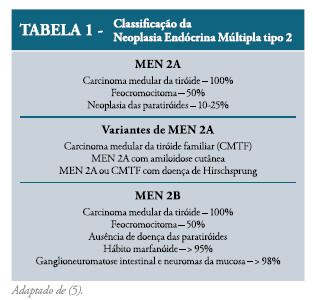
A MEN 2A, ou síndrome de sipple, compreende 75% dos casos da síndrome MEN tipo 2 e caracteriza-se pela presença de carcinoma medular da tiróide (CMT), feocromocitoma e neoplasia das paratiróides. Estão descritas variantes da síndrome: carcinoma medular da tiróide familiar (CMTF), MEN2A com amiloidose cutânea e MEN 2A ou CMTF com doença de Hirschsprung.1
A associação de CMT, feocromocitoma, múltiplos neuromas da mucosa, hábito marfanóide e ausência de doença das paratiróides classifica-se como MEN 2B.5
Síndromes clínicas
Carcinoma medular da tiróide
O CMT é um tumor produtor de calcitonina (CT), com origem nas células C ou parafoliculares da tiróide. Cerca de 4% dos carcinomas da tiróide são carcinomasmedulares,sendoque75%ocorremdeforma esporádica e os restantes 25% são hereditários.6 dos CMT hereditários, a maioria desenvolve-se como parte integrante da síndrome MEN 2A (cerca de 80%), enquanto os restantes ocorrem sob a forma de CMTF (10 a15%) ou associam-se à síndrome MEN 2B (5 a 10%).4
Todas as variantes de MEN2 têm uma penetrância elevada de CMT, de cerca de 100%, sendo a primeira manifestação da síndrome na maioria dos casos. É uma neoplasia multicêntrica e geralmente bilateral, que apresenta como lesão precursora a hiperplasia multifocal de células C. Progressivamente, ao longo dos anos, a hiperplasia de células C progride para hiperplasia nodular, CMT microscópico e, finalmente, CMT macroscópico. As lesões são frequentemente multifocais, achado que se pensa representar a expansão de células clonais individuais e não metástases intratiroideias.5
Apresenta um comportamento biológico variável. Pode metastizar para os gânglios linfáticos ou à distância, sobretudo para o fígado, pulmões e ossos. A metastização linfática é rara se houver apenas hiperplasia de células C. No entanto, mais de 80% dos tumoressuperioresa1cmapresentammetástasesem gânglios linfáticos locais.1 Em alguns doentes, a ocorrência de diarreia pode ser a primeira manifestação clínica. Embora a etiologia esteja pouco esclarecida, crê-se que seja causada por um factor humoral produzido pelo tumor.7 outra manifestação associada ao CMT é a síndrome de secreção ectópica de ACTH. É uma síndrome rara, observada mais frequentemente em doentes com tumores primários de grandes dimensões ou com lesões metastáticas.8
A CT é o principal produto de secreção do CMT.
O valor basal de CT correlaciona-se geralmente com a massa tumoral, estando habitualmente aumentado em doentes com tumores palpáveis. Em doentes com tumores pequenos ou com hiperplasia de células C, o doseamento basal pode ser normal.9 A CT também pode ser doseada após provas de estimulação com pentagas trina ou com cálcio. Pode haver aumento dos valores de CT (basais e após estimulação) em doentes com tiroidite auto-imune, insuficiência renal, tumores neuroendócrinos ou anticorpos heterofílicos.6 Em algumas famílias, a ocorrência de resultados falsos positivos chega a ser de 15% do número total de portadores.1 desde o aparecimento dos testes genéticos, a determinação de CT tem maior relevância como marcador tumoral. Permite avaliar a progressão da doença a longo prazo e é um bom indicador da agressividade do tumor na doença metastática. Após cirurgia, valores elevados geralmente indicam doença persistente ou recorrente.10 Nos últimos anos, houve uma melhoria da sensibilidade dos métodos de doseamento de CT basal e as provas de estimulação têm sido cada vez menos recomendadas. Actualmente considera-se que, após cirurgia, valores de CT aumentados apenas nas provas de estimulação indicam que o volume de doença residual é baixo, sendo improvável a sua detecção em exames imagiológicos bem como a sua resolução com terapêuticas adicionais.6 Além da CT, o CEA (CarcinoEmbryonic Antigen) também pode ser utilizado como marcador tumoral.
O tratamento de primeira linha destes tumores é cirúrgico, devendo ser realizado o mais precocemente possível. O procedimento primário consiste na tiroidectomia total com dissecção dos gânglios linfáticos da cadeia central. se houver evidência de envolvimento ganglionar cervical lateral, é necessária uma dissecção mais alargada.10 Após a primeira intervenção cirúrgica, se o doseamento de CT for elevado, é necessário determinar a extensão de doença metastática local e à distância. Se houver suspeita ou confirmação de doença metastática local (cervical e/ ou mediastino superior) sem evidência de metástases à distância, o procedimento defendido é a reintervenção cirúrgica.4 Estudos baseados em centros de larga experiência relatam que, nestes casos, cerca de 5 a 15% dos doentes apresentam valores de CT normais ou indetectáveis após a segunda cirurgia.1 se houver evidência de metástases à distância, não há indicação para reintervenção cirúrgica, excepto se o doente desenvolver diarreia.4 o CMT é um tumor pouco sensível à radiação. A radioterapia pode ser utilizada com intenção paliativa para diminuir a carga tumoral cervical e para prevenir a recorrência local. Contudo, permanecem questionáveis os seus benefícios em termos de morbilidade e mortalidade.11 os esquemas de quimioterapia não têm benefício demonstrado no CMT metastático, estando apenas indicados em casos de rápida progressão tumoral.4 Os análogos da somatostatina não devem ser considerados, dado que, neste contexto, são ineficazes no controlo do crescimento tumoral. Na ausência concomitante de carcinoma diferenciado da tiróide com origem nas células epiteliais foliculares, a terapêutica com iodoradioactivo não está recomendada.6
Feocromocitoma
O feocromocitoma é uma neoplasia das células cromafins da suprarrenal, que, no contexto da síndrome MEN tipo2, afecta cerca de 50% dos portadores.12 Em termos histológicos, o tecido cromafim apresenta o mesmo tipo de progressão que as células C, incluindo hiperplasia, expansão difusa e, final-mente, feocromocitoma. o padrão habitual é de multicentricidade num fundo de hiperplasia medulardifusa.1 Pode ser unilateral ou bilateral, encontrase geralmente limitado à suprarrenal e raramente é maligno. Observa-se frequentemente invasão capsular, mas tal achado não apresenta correlação com a recorrência do tumor.5
Os feocromocitomas associados à síndrome MEN tipo 2 apresentam frequentemente um excesso de produção relativo de adrenalina, o que, clinicamente, se traduz por uma baixa incidência de hipertensão e pelo predomínio de sintomas ß-adrenérgicos, como palpitações, taquicardia e nervosismo. Em doentes com feocromocitomas de maiores dimensões também há a produção excessiva de noradrenalina, embora a razão adrenalina/noradrenalina permaneça elevada. Nestes casos, a hipertensão pode ser um problema clínico importante.5
O doseamento das catecolaminas e metanefrinas fraccionadas plasmáticas apresenta elevada sensibilidade para o diagnóstico de feocromocitoma. o doseamento das catecolaminas e metanefrinas urinárias é actualmente menos utilizado. A excreção urinária de ácido vanilmandélico é habitualmente normal na fase inicial da doença, apresentando utilidade limitada para a detecção do tumor.5
O diagnóstico é confirmado por TAC (Tomografia Axial Computorizada) ou por RM (Ressonância Magnética) abdominal. A TAC permite obter uma melhor resolução anatómica e é menos dispendiosa. A RM tem maior especificidade e pode ser útil para distinguir um feocromocitoma pequeno de um adenoma do córtex da suprarrenal.1 A cintigrafia com metaiodobenzilguanidina i131 (MIBG) permite confirmar a presença de tecido cromafim funcionante e excluir localização ectópica. No entanto, não é útil para distinguir hiperplasia suprarrenal de feocromocitoma. Recentemente, a imagiologia funcional com 18F-fluorodopamina PET (Positron Emission Tomography) tem sido utilizada como alternativa ao MIBG no estudo de localização dos feocromocitomas e parece ser particularmente útil em doentes com doença recorrente.13
O tratamento de eleição do feocromocitoma é cirúrgico e, quando presente, deve ser o primeiro tumor a ser tratado cirurgicamente. Antes da sua remoção, está indicado tratamento farmacológico com antagonistas a-e/ou ß-adrenérgicos e/ou a-metiltirosina.4 A adrenalectomia laparoscópica unilateral é o procedimento de escolha para o feocromocitoma unilateral.5 A adrenalectomia bilateral está indicada quando ambas as glândulas apresentam o tumor. A insuficiência suprarrenal é um importante efeito indesejável nos doentes submetidos à cirurgia bilateral, pelo que, nos últimos anos, tem sido aplicada uma técnica que tenta preservar o tecido cortical, designada por adrenalectomia parcial. Em pequenos grupos de doentes, permitiu a manutenção da função da suprarrenal em cerca de 80% dos casos.1 A taxa de recorrência descrita é de cerca de 20%. Embora a experiência seja ainda limitada, deve ser considerada quando a manutenção da função cortical for considerada imperativa.6
Hiperparatiroidismo
O hiperparatiroidismo (HPT) primário é uma manifestação mais tardia da doença, ocorrendo em 10 a 25% dos doentes com a síndrome MEN 2A. Na maioria dos casos é assintomático, embora possa ocorrer litíase renal, osteíte fibrosa quística, doença ulcerosa péptica e pancreatite.4 As indicações para intervenção cirúrgica são apresentadas na Tabela 2
, sendo idênticas às do HPT primário esporádico.1 A realização de estudos de localização pré-operatória (como ecografia, cintigrafia com sestamibi e/ou TAC cervical e torácica) está recomendada em doentes com cirurgia cervical prévia por CMT e com doença recorrente ou persistente.11 Em doentes submetidos a tiroidectomia total por hiperplasia de células C detectada precocemente, a incidência de HPT é muito baixa. Por este motivo, a maioria dos cirurgiões opta por remover apenas as paratiróides que se encontram aumentadas aquando da cirurgia. Nos raros casos em que o HPT primário é uma manifestação proeminente da síndrome deve considerar-se a realização de paratiroidectomia total com auto transplante no antebraço não dominante.5

Carcinoma medular da tiróide familiar
O CMTF é uma variante clínica da síndrome MEN 2A em que o CMT é a única manifestação clínica.5 o seu diagnóstico requer a presença de mais de dez portadores na família e de vários portadores com idade superior a 50 anos. Em famílias pequenas ou apenas com uma geração afectada, recomenda-se cautela na classificação de CMTF devido à possibilidade de síndrome MEN 2A e risco associado de feocromocitoma.14
Men 2A com amiloidose cutânea
Pelo menos 15 famílias com a síndrome MEN 2A têm associada uma lesão cutânea pruriginosa, unilateral ou bilateral, na região escapular. Na maioria dos doentes, o aparecimento de prurido intermitente precede o desenvolvimento da lesão em 3 a 5 anos. Histologicamente, caracteriza-se pela deposição de substância amilóide na junção dermo-epidérmica.5
Men 2A com doença de Hirschsprung
A variante da síndrome MEN 2A associada a doença de Hirschsprung, embora rara, pode ocorrer devido à ausência de células ganglionares autonómicas no plexo parassimpático do cólon distal.6 Pode ser diferenciada da ganglioneuromatose associada à síndrome MEN 2B por biópsia rectal.1
Outras manifestações da síndrome Men 2B
A associação de CMT, feocromocitoma, múltiplos neuromas da mucosa e hábito marfanóide, sem doença das paratiróides, classifica-se como MEN 2B. A síndrome MEN 2B é a forma mais rara e agressiva de MEN tipo 2 com o desenvolvimento de CMT em idades mais precoces.6 Está descrita a ocorrência de doença metastática em crianças com idade inferior a 1 ano.1 o feocromocitoma afecta cerca de 50% dos portadores e ocorre em faixas etárias semelhantes e com manifestações idênticas às da síndrome MEN 2A.5 A principal característica fenotípica da doença é a presença de múltiplos neuromas da mucosa na porção distal da língua, lábios e áreas subconjuntivais. os neuromas também podem existir no tracto gastrointestinal e causar flatulência, dor abdominal e obstrução ou pseudo-obstrução intestinal. outras manifestações associadas à síndrome incluem hábito marfanóide, pectus excavatum, deslocação das epífises femorais e hiperextensão das articulações.1
Tratamento
Genética
O proto-oncogene RET (rearranjed during transfection) é o gene responsável pela síndrome MEN tipo2,tendosidodescobertoem1985.ogeneRET localiza-se no cromossoma 10, é constituído por 21 exões e codifica um receptor do tipo tirosina cinase, que é formado por um domínio extracelular, uma região transmembranar e um domínio intracelular tirosina cinase.15 A importância clínica do RET na oncologia foi pela primeira vez reconhecida no carcinoma papilar da tiróide. Em 25 a 35% dos carcinomas papilares da tiróide são identificadas mutações somáticas do RET que provocam um rearranjo genético com a fusão de dois genes diferentes (como o oncogene RET-PTC). Pelo contrário, a síndrome MEN tipo 2 resulta de mutações germinativas do RET que causam ganho de função da proteína rET.4 A proteína rET parece estar relacionada com a transdução de sinais de crescimento e diferenciação em vários tecidos incluindo os que têm origem da crista neural.9
Indicações para o estudo genético
A síndrome MEN tipo 2 é um dos exemplos em que o estudo genético tem importantes implicações na intervenção clínica, de tal forma que a decisão de efectuar tiroidectomia é baseada na análise genética das mutações do proto-oncogene RET e não nodoseamento da CT.16 A tiroidectomia profilática com base no estudo genético justifica-se, porque a detecção e intervenção precoces podem alterar a evolução do CMT, a tiroidectomia é bem tolerada na maioria dos casos, o doseamento de CT tem uma percentagem importante de falsos positivos e a análise genética tem uma percentagem elevada de verdadeiros positivos e poucos falsos negativosefalsospositivos.4
Em cerca de 99% dos casos MEN 2 identifica-se um amutação germinativa do RET. A análise de três exões (10, 11 e 16) permite a identificação de mais de 95% das mutações. A pesquisa adicional dos exões 13, 14 e 15 aumenta a proporção de mutações identificadasparamaisde98%.seapesquisainicial for negativa, os restantes exões devem ser sequenciados. Devido à possibilidade de resultados falsos positivos ou falsos negativos, é importante repetir a análise genética numa amostra de ADN obtida de uma colheita sanguínea diferente da anterior.5 Se o resultado se mantiver negativo mas a suspeita clínica for elevada, devem ser considerados testes de ligação génica do locus RET.4
As indicações para pesquisa de mutações germinativas do RET são apresentadas na Tabela 3
. Todos os casos de CMT devem ser testados.1 Cerca de 1 a 7% dos CMT aparentemente esporádicos apresentam mutações do gene RET, das quais cerca de 2 a 9% são mutações germinativas de novo.6 A definição de feocromocitoma aparentemente esporádico inclui a presença de doença unilateral, história familiar negativa e ausência de sinais ou sintomas de MEN tipo 2. A pesquisa de mutações do RET está indicada em doentes com feocromocitoma que não cumpram esses critérios.14 Nos casos de HPT aparentemente esporádico, sem suspeita clínica de MEN tipo 2, o estudo genético não está indicado.1

Implicações terapêuticas do estudo genético
O codão do RET que se encontra especificamente mutado correlaciona-se com a variante da síndrome e com a agressividade do CMT. As mutações do RET podem ser estratificadas em quatro categorias (Tabela 4
), que se referem ao risco de metastização local e à distância em idade precoce.6,11
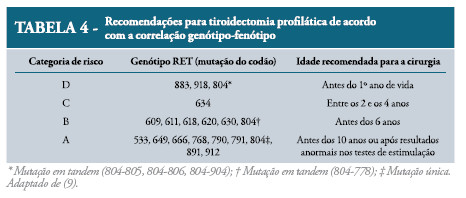
Risco muito elevado (D) – Crianças com predisposição para a síndrome MEN 2B, que está frequentemente associada a mutações dos codões 883, 918 e mutações em tandem do codão 804 (804-805, 804-806, 804-904). recomenda-se a realização de tiroidectomia total com dissecção dos gânglios da cadeia central no primeiro ano de vida. Nestes doentes, a presença de CMT microscópico é comum durante o primeiro ano de vida, estando também descrita a ocorrência de metástases à distância. A intervenção cirúrgica em idades mais avançadas é frequentemente não curativa.
II. Risco elevado (C) – Crianças com mutações do codão 634 têm indicação para tiroidectomia total entre os 2 e os 4 anos de idade. A dissecção dos gânglios centrais reúne menos consenso. A maioria dos cirurgiões opta pela dissecção ganglionar aquando do primeiro procedimento cirúrgico, devido à maior morbilidade associada a uma intervenção subsequente.
III. Risco intermédio (B) – doentes com mutação dos codões 609, 611, 618, 620, 630 e mutações em tandem do codão 804 (804-778) devem ser submetidos a tiroidectomia total antes dos 6 anos de idade.
IV. Risco baixo (A) – doentes com mutações dos codões 533, 649, 666, 768, 790, 791, 804 (mutação única), 891 ou 912 podem ser submetidos a tiroidectomia total até aos 10 anos de idade ou quando houver valores de CT anormais em testes de estimulação efectuados periodicamente.
Monitorização dos tumores associados à síndrome Men tipo 2
Após realização da tiroidectomia, devem ser realizados doseamentos de CT e CEA dentro de 2 a 3 meses. se o doseamento pós-operatório de CT basal for indetectável, o risco de doença residual persistente ou recorrente é baixo e pode ser iniciada vigilância regular. A realização de outros testes ou exames imagiológicos não é mandatória, embora possa ser realizada uma ecografia cervical de base.6 Em doentes com doseamentos de CT detectáveis, as medidas de vigilância recomendadas dependem do valor de CT. Doentes com CT <150 pg/ml devem realizar ecografia cervical e pode ser considerada a realização de exames imagiológicos adicionais para servirem de estudo base. No entanto, esses exames podem ser protelados e subsequentemente implementados se houver aumento posterior da CT. Doentes com CT =150 pg/ml devem realizar ecografia cervical e exames imagiológicos adicionais para localização de metástases à distância.6 Em todos os portadores é importante que se faça uma revisão anual das manifestações clínicas sugestivas de feocromocitoma.5,9 A presença de sinais ou sintomas associados a excesso de catecolaminas ou massa suprarrenal requer avaliação bioquímica imediata de feocromocitoma. Na ausência de sintomas ou alterações analíticas sugestivas, o rastreio do tumor através de exames imagiológicos abdominais não está recomendado. O rastreio bioquímico deve realizar-se anualmente e iniciar-se aos 8 anos de idade nos portadores de mutações associadas à síndrome MEN 2B e de mutações dos codões 630 e 634, e aos 20 anos de idade nos portadores de outras mutações associadas à síndrome MEN 2A.4,6
O rastreio de HPT primário deve realizar-se anualmente e incluir doseamentos séricos de cálcio ionizado ou cálcio corrigido com albumina (com ou sem doseamentos séricos de paratormona intacta).
A monitorização deve iniciar-se aos 8 anos de idade nos portadores de mutações dos codões 630 e 634 e aos 20 anos de idade nos portadores de outras mutações associadas à síndrome MEN 2A.4,6
Perspectivas futuras
A identificação de mutações activadoras do protooncogene RET como a causa de mais de 99% dos CMT hereditários e de cerca de 25% dos CMT esporádicos tem levado ao desenvolvimento de terapêuticas dirigidas à inactivação da proteína RET.5
Fármacos designados por inibidores da tirosina cinase têm sido utilizados em ensaios clínicos com alguma eficácia, sobretudo em doentes com doença localmente avançada ou metastática.17 Recentemente têm sido publicados resultados de estudos em fase 1 e 2 que mostram taxas de resposta parcial (redução do tamanho do CMT em mais de 30%) de aproximadamente 2% para o motesanib, 20% para o vandetanib, 44% para o sorafenib e 41% para o XL-184.5 Embora ainda se desconheça se o uso destes agentes altera o curso da doença, os dados publicados sugerem que podem constituir uma componente promissora na abordagem terapêutica.
Referências
1. Marx Sj, Wells Sa. Multiple Endocrine Neoplasia. In: Williams Textbook of Endocrinology. 12th Ed. Elsevier; 2011. P. 1743-55. [ Links ]
2. Marsh Dj, Zori Tr. Genetic Insights Into Familial Cancers – Update and Recent Discoveries. Cancer Lett 2002;181(2):125-64. [ Links ]
3. Scriver Cr, Beaudet Al, Sly Ws, Valle D. The Metabolic and Molecular Bases of Inherited Disease. In: Multiple Endocrine Neoplasia Type 2. 8th Ed. New York: Mcgraw Hill; 2001. P. 931-42. [ Links ]
4. Brandi Ml, Gagel Rf, Angeli A, et al. Guidelines for Diagnosis and Therapy of MEN Type 1 and Type 2. J Clin Endocrinol Metab 2001;86(12):5658-71. [ Links ]
5. Jameson Jl, De Groot Lj. Endocrinology – Adult and Pediatric. 6th Ed. Elsevier; 2010. [ Links ]
6. Kloos Rt, Charis E, Douglas BE, et al. Medullary Thyroid Cancer: Management Guidelines of The American Thyroid Association. Thyroid 2009;19(6):565-612. [ Links ]
7. Isaacs P, Whittaker Sm, Turnberg La. Diarrhea Associated With Medullary Carcinoma of The Thyroid. Gastroenterology 1974;67:521-6. [ Links ]
8. Deftos Lj, Murray Ss, Burton Dw, et al. A Cloned Chromogranin A (Cga) Cdna Detects A 2.3Kb Mrna In Diverse Neuroendocrine Tissues. Biochem Biophys Res Commun 1986;137(1):418-23. [ Links ]
9. Lips CJ. Classification and Genetics of Multiple Endocrine Neoplasia Type 2. 2011 Set. Disponível Em Url: http://www.uptodate.com [ Links ]
10. Guimarães J. Multiple Endocrine Neoplasia. Acta Med Port 2007;20(1):65-72. [ Links ]
11. Lips CJ. Approach To Therapy In Multiple Endocrine Neoplasia Type 2. 2011 Set. Disponível Em Url: http://www.uptodate.com [ Links ]
12. Almeida MQ, Stratakis CA. Solid Tumors Associated With Multiple Endocrine Neoplasias. Cancer Genet Cytogenet 2010;203(1):30-6. [ Links ]
13. Pacak K, Eisenhofer G, Ilias I. Diagnosis of Pheochromocytoma With Special Emphasis On MEN2 Syndrome. Hormones 2009;8(2):111-6. [ Links ]
14. Lips CJ. Clinical Manifestations and Diagnosis of Multiple Endocrine Neoplasia Type 2. 2011 Set. Disponível Em Url: http://www.uptodate.com [ Links ]
15. Marini F, Falchetti A, Del Monte F, et al. Multiple Endocrine Neoplasia Type 2. Orphanet J Rare Dis 2006;1:45. [ Links ]
16. Raue F, Frank-Raue K. Genotype-Phenotype Relationship In Multiple Endocrine Neoplasia Type 2. Implications for Clinical Management. Hormones 2009;8(1):23-8. [ Links ]
17. Lodish M, Stratakis C. Ret Oncogene In MEN2, MEN2B, MTC and Other Forms of Thyroid Cancer: Molecular Genetics and Therapeutic Advances. Expert Rev Anticancer Ther 2008;8(4):625-32. [ Links ]
18. Bringhurst Fr, Demay MB, Kronenberg Hm. Hormones and Disorders of Mineral Metabolism. In: Williams. Textbook of Endocrinology. 12th Ed. Elsevier; 2011;1237-304. [ Links ]
Pedro Rodrigues
Serviço de Endocrinologia, Diabetes e Metabolismo Centro Hospitalar de São João E.P.E. Alameda Prof. Hêrnani Monteiro 4200-319 Porto. Email: pmm_rodrigues@hotmail.com

