








Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArquivos de Medicina
On-line version ISSN 2183-2447
Arq Med vol.23 no.6 Porto Nov. 2009
Troponina
Estrutura, Fisiopatologia e Importância Clínica para Além da Isquemia Miocárdica
Carla Sofia Martins
Serviço de Medicina Interna, Hospital de São João, Porto
As troponinas cardíacas (I e T), introduzidas, na definição de enfarte agudo do miocárdio (EAM), inicialmente em 2000, pelas directrizes conjuntas da American College of Cardiology (ACC) e da European Society of Cardiology (ESC), e actualizadas em 2007, são consideradas, actualmente, os biomarcadores bioquímicos padrão de necrose miocárdica, dada a sua cardio-selectividade e ratio sinal / ruído elevado (o ruído depende do limiar de detecção da troponina, que com os testes actuais é muitíssimo baixo).
A ânsia de diagnosticar enfarte do miocárdio nas primeiras 4h do início dos sintomas, levou ao aperfeiçoamento dos testes de doseamento da troponina (Tp), de tal forma que, presentemente, é possível detectar níveis extremamente baixos dessa proteína no plasma, com a consequência inevitável do aparecimento de indivíduos (incluindo pessoas saudáveis) com níveis detectáveis de Tp cujo significado fica por esclarecer. Uma possível interpretação é que essa Tp resulte de lesão, não isquémica, dos miocardiócitos, subsequente a certas agressões hemodinâmicas / químicas, como a hipoxia, a hipertensão arterial sistémica ou pulmonar, a insuficiência renal ou a hipertrofia ventricular esquerda, entre outros.
Palavras-chave: troponina cardíaca; isquemia; hipoxia.
Troponins
Cardiac troponins (I and T), introduced, into acute myocardial infarction definition, formerly in 2000, by the joint task force of American College of Cardiology (ACC) and European Society of Cardiology (ESC), and updated in 2007, are considered, nowadays, the standard chemical biomarkers of myocardial necrosis, in face of their cardio-selectivity and high sign/noise ratio (the noise depending on troponina detection threshold, which is actually very low with current assays).
The urge to diagnose myocardial infarction in the first 4h of symptoms presentation conducted to improvements in troponin assays, in such a way that currently is possible to detect extremely low levels of that protein in plasma, which leads inevitably to appearance of people (including those formerly named healthy) with detectable levels of troponina, whose meaning remains to be clarified. One possible explanation is that this troponina results from non-ischaemic myocardiocytes injury, in response to certain homodynamic/chemical stress, like hypoxia, systemic or pulmonary hypertension, renal failure or left ventricular hypertrophy, among others.
Key-words: cardiac troponins; ischaemia; hypoxia.
1 - INTRODUÇÃO
A troponina I foi inicialmente considerada um biomarcador proteico de necrose miocárdica, tendo nos últimos anos, sido encarada como a análise bioquímica padrão para o diagnóstico de enfarte agudo do miocárdio, designadamente do enfarte sem supra desnivelamento do segmento ST (EMSST), a par dos critérios clínicos e electrocardiográficos / angiográficos.
Pela cinética da libertação da troponina I a partir dos cardiócitos lesados, doentes com enfarte agudo do miocárdio podem apresentar valores normais de TpI no sangue, nas primeiras horas após o evento. De facto, os primeiros testes de doseamento da troponina só mostravam subida da TpI 4h a 6h depois, com as inerentes implicações no diagnóstico e prognóstico, dado o benefício do diagnóstico e intervenção precoces.
Por este motivo, os fabricantes aperfeiçoaram os seus testes tornando-os cada vez mais sensíveis, permitindo a detecção precoce de níveis extremamente baixos de TpI no sangue, e portanto o diagnóstico mais atempado do enfarte agudo do miocárdio. As vantagens são óbvias, mas existe um preço a pagar. De facto, dispondo de testes com limiar de detecção de TpI mais baixo, indivíduos previamente tidos como saudáveis por apresentarem valores encarados como normais, serão agora considerados doentes ou pelo menos portadores de TpI detectável, não sendo claro o seu significado patológico.
Ultimamente tem-se investigado amplamente esta questão, e os estudos são unânimes em demonstrar que pequenas elevações da TpI plasmática, ainda que abaixo do percentil 99 (usado actualmente como valor discriminativo para diagnóstico de enfarte agudo do miocárdio), têm significado prognóstico, particularmente quanto à ocorrência de eventos cardiovasculares adversos e à sobrevida. E isto verifica-se não só em doentes com doença cardíaca isquémica estável como também numa série de outras situações patológicas, nomeadamente em doentes críticos, na insuficiência cardíaca, no tromboembolismo pulmonar, nainsuficiência renalcrónica, na endocardite infecciosa, na doença cardíaca valvular e na doença pulmonar crónica obstrutiva (DPOC).
Se é consensual que níveis detectáveis de TpI têm implicações significativas no prognóstico dos doentes, o mesmo não se passa relativamente à controvérsia gerada pelas tentativas de explicação da sua etiologia.
2 - CORAÇÃO: BOMBA MUSCULAR
O coração é o operador central de todo o sistema cardiovascular, consistindo em duas poderosas bombas musculares (coração direito e coração esquerdo), que em cada ciclo cardíaco bombeiam o sangue e o distribuem por todo o organismo.
Cada fibra muscular cardíaca constitui-se por miofibrilas (1), células, em forma de Y, que se encontram separadas umas das outras por comunicações altamente permeáveis (as “gap-junctions”), de tal forma que, funcionalmente, se podem considerar verdadeiros sincícios celulares (Figura 1). De facto, existem dois sincícios celulares distintos – auricular e ventricular – separados entre si por tecido fibroso. Em cada miofibrila, por sua vez, encontram-se milhares de filamentos de actina (finos) e miosina (grossos), em íntima relação uns com os outros.

Fig. 1 - Fibra muscular cardíaca – constituída por várias células em forma de Y. O aspecto estriado deve-se à alternância de filamentos finos (mais claros) e grossos (mais escuros). Os traços mais claros que separam as células são os discos intercalados – folheto duplo de membrana celular, onde se encontram as “gap-junctions”.
Adaptado de http://www.visualhistology.com/products/atlas/VisualHistology_Atlas_2-0-09_1.jpg)
Contrariamente ao músculo esquelético, o músculo cardíaco não se contrai em resposta à estimulação nervosa (controlada pelo Sistema Nervoso Central), dependendo da auto-excitabilidade e miogenicidade do nó sinusal, o pace-maker cardíaco (regulado pelo Sistema Nervoso Autónomo).
Quando uma célula muscular é excitada, o potencial de acção difunde-se para as outras células vizinhas (pelas “gap-junctions”), deslocando-se das aurículas para os ventrículos, através de células especializadas na condução nervosa (células de Purkinje), de forma a garantir a sua contracção ritmada e harmoniosa, em cada ciclo cardíaco.
3 -CONTRACÇÃO DO MIOCÁRDIO: ACOPLAMENTO EXCITAÇÃO-CONTRACÇÃO
Após a excitação nervosa, o cálcio é estimulado a sair do retículo sarcoplasmático, para o sarcoplasma, onde se encontram os miofilamentos, e é da interacção do cálcio comlocais específicos desses miofilamentos, como veremos adiante, que resulta o deslizar relativo da actina e miosina, e a subsequente contracção muscular.
3.1 - Miofilamentos: estrutura, fisiologia e regulação pelo cálcio
No músculo cardíaco, como em todo o músculo estriado, a activação pelo cálcio é regulada mormente ao nível dos miofilamentos finos.
Concretamente, os filamentos finos (2) são constituídos por dois polímeros helicoidais de actina filamentosa, entrançados em duas cadeias de tropomiosina (Figura 2), cada uma ligada a um complexo troponínico, que, por sua vez, é formado por três subunidades – troponina C, troponina I e troponina T – na razão molar de 1:1:1. As tropomiosinas de cada filamento comunicam com as dos filamentos adjacentes, por interacções topo-a-topo, parecendo fundamentais à difusão da onda de excitaçãocontracção, ao longo de toda a fibra muscular (3). Além disso, são proteínas cruciais na regulação da interacção entre os filamentos de actina e miosina. De facto, con-forme a posição dessas proteínas relativamente à actina, os miofilamentos finos podem adoptar um de três modos de estado (2,4):
• Modo B – Também designado por modo bloqueado, porque neste modo de estado, os filamentos finos estão bloqueados ao estabelecimento de qualquer ponte cruzada entre a actina e a miosina, não podendo ocorrer contracção muscular. É um tipo de conformação espacial que vigora em situações de baixa concentração de cálcio sarcoplasmático, como é o caso das situações de repouso muscular.
• Modo C – Também designado por modo cálcioactivado. Decorre da libertação do cálcio a partir do retículo sarcoplasmático para o sarcoplasma, após excitação celular. Subsequentemente, esse cálcio liga-se à troponina C, fazendo com que o efeito inibitório da troponina I se atenue (ver adiante), e a tropomiosina (5) se desloque a partir dos sulcos de actina, deixando a descoberto o domínio S1 da actina, onde se vai ligar a miosina, culminando com o estabelecimento de pontes cruzadas entre ambos os filamentos. Porém, e apesar da formação destas pontes cruzadas, não se produz contracção muscular, por serem, ainda, ligações fracas.
• Modo M – Também designado por modo aberto ou estado induzido pela miosina. Neste modo estabel-ecem-se pontes altamente potentes (na realidade trata-se apenas de uma (6) ponte cruzada potente) entre os filamentos de actina e os de miosina, pelo efeito activador da actina sobre a ATP-ase (adenosina trifosfatase) da miosina, produzindo-se força muscular eficaz.
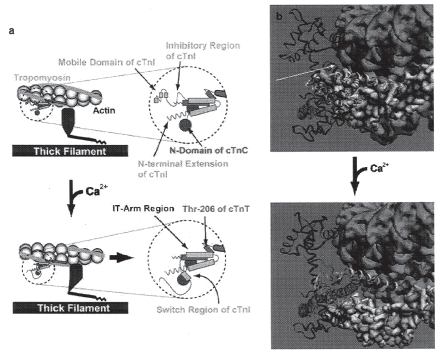
Fig. 2 - Miofilamentos de actina e miosina, e o papel do cálcio na sua regulação.(adaptado de Pflugers Arch -Eur J Physiol (2008) 457:37–46)
O papel regulador do cálcio assenta, portanto, na alteração do equilíbrio entre aqueles três modos de estado dos miofilamentos finos. No músculo cardíaco estima-se que, na ausência de cálcio, cerca de 50% das unidades reguladoras dos filamentos finos se encontrem no modo B e 40% no modo C, passando aproximadamente 75% ao modo C quando esse catião está presente (5,7).
O cálcio, porém, não actua de forma isolada, reconhecendo-se 2 factores essenciais ao seu efeito regularizador da conformação dos miofilamentos e contracção muscular: 1 – quantidade de cálcio libertada a partir do retículo sarcoplasmático; e 2 – resposta (sensibilidade) dos miofilamentos ao cálcio.
Por sua vez, o comprimento dos sarcómeros (relação de Frank-Starling) e a modificação pós-translacional das proteínas sarcoméricas, designadamente a fosforilação dependente da cínase, constituem os principais factores moduladores da sensibilidade ao cálcio. Um exemplo concreto é a interacção topo-a-topo das tropomiosinas, já referido atrás, que se descobriu recentemente (2), ser controlado pela fosforilação do resíduo Ser-238 dessas proteínas.
3.2 - Troponinas
De acordo com o exposto anteriormente, as troponinas são proteínas reguladoras que controlam a interacção Ca2+-dependente entre os filamentos de actina e miosina, sendo responsáveis pelo ciclo dinâmico de contracção e relaxamento muscular. Reconhecem-se três troponinas distintas (2), designadas pelas letras C, T e I.
A troponina C (TpC) é, por excelência, a proteína de ligação (reversível) ao cálcio, que, uma vez activada por ele, permite a mudança de conformação dos miofilamentos finos em modo B para o modo C (ver atrás). A molécula da troponina C é dotada de dois domínios fundamentais, unidos por um ligante central – terminais N e C – dispondo de dois pontos de ligação ao cálcio, em cada um desses domínios, denominados locais I e II no primeiro e III e IV no segundo. Estes dois últimos locais mostram grande afinidade para o cálcio, pelo que não podem controlar a contracção muscular dependente desse ião. Por este motivo, houve (2) quem classificasse o domínio C como sendo estrutural. O local I do domínio N, por seu turno, não se pode ligar ao cálcio, em condições fisiológicas, devido à substituição de aminoácidos-chave. Assim, apenas o local II deste mesmo domínio, constitui o ponto crucial de ligação ao cálcio.
Nesta sequência, percebe-se que a contracção do músculo cardíaco é regulada apenas por um ponto de ligação ao cálcio, contrariamente ao músculo esquelético, onde funcionam efectivamente dois pontos de ligação.
Não obstante, o cálcio só conduz à contracção muscular após ligação à troponina C se esta estiver integrada num miofilamento fino, levando a supor que exista um mecanismo de feedback positivo transmitido ao longo dos diversos filamentos, realçando, uma vez mais, a importância da interacção entre os topos das tropomiosinas.
A troponina I, um monómero com 23,5 kDa, corresponde à componente inibitória do complexo troponínico, inibindo, portanto, a contracção muscular quando a concentração do cálcio plasmático é baixa. Tal como a troponina C, ou qualquer outra proteína, a troponina I tem um terminal N (domínio inibitório) e um terminal C (domínio de ligação à actina). A ligação da actina ao domínio inibitório, faz com que a troponina I iniba o efeito activador da actina sobre a ATP-ase (adenosina trifosfatase) da miosina, conduzindo ao relaxamento muscular. Pensa-se que o terminal C esteja intimamente envolvido na indução da mobilização da tropomiosina para uma posição inibitória que bloqueie a interacção entre a miosina e a actina.
O domínio inibitório tem uma sequência particular de aminoácidos que se tem mantido preservada ao longo da evolução das espécies, sugerindo tratar-se de um ponto crítico na regulação do ciclo contracção / relaxamento muscular cardíaco. De facto, já foram documentadas alterações nessa sequência de aminoácidos, conducentes a um defeito da acção inibitória da troponina I durante a diástole, em certos tipos de insuficiência cardíaca (IC).
Entre os terminais C e N da troponina I, existe uma zona intermédia – Switch zone – onde se liga a troponina C. Relembremos que após ligação do cálcio à troponina C, esta seliga à zonaintermédia da troponina I, obrigando ao deslizar dos seus domínios N e C, esmorecendo o seu efeito inibitório sobre a actina.
O terminal N (T1) da troponina T (TpT) liga-se fortemente à tropomiosina (donde deriva o seu nome), estando estrategicamente situado na zona de interface entre os topos das tropomiosinas, pensando-se que regule, precisamente, as interacções topo-a-topo, já mencionadas anteriormente neste texto. O terminal C (T2) da troponina T liga-se às troponinas I e C, servindo de intermediário entre ambas. Presentemente, supõe-se que a fosforilação da troponina T cardíaca tenha um papel significativo na regulação do miofilamento fino cardíaco.
4 -TROPONINA I: ESPECIFICIDADES DA MOLÉCULA E IMPORTÂNCIA CLÍNICA
Conhecem-se trêsisoformas da troponina I –cardíaca, esquelética lenta e esquelética rápida – cada uma codificada por um gene específico.
No coração embrionário dos mamíferos, as células musculares exprimem predominantemente a forma esqueléticalenta da troponina I. Teoriza-se (8) que esta forma seja fundamental para a robustez da bomba muscular cardíaca, quando da adaptação à hipoxia associada ao parto, pelo facto de aumentar a sensibilidade ao cálcio bem como o tempo de relaxamento e a resistência a alterações do pH, e diminuir a sensibilidade à estimulação β-adrenérgica. Todavia, após o parto, a expressão dessa isoforma é drasticamente reduzida, e na idade adulta apenas ocorre a forma cardíaca, salvo em certas situações patológicas como é o caso da IC.
Nos últimos anos, sobretudo após a redefinição dos critérios de diagnóstico do enfarte agudo do miocárdio, em que se enalteceu o papel da troponina, esta proteína tem sido alvo de estudo intenso, na expectativa de se compreender melhor a sua estrutura e patofisiologia.
Efectivamente, existe já evidência (9, 10) que certas alterações estruturais da isoforma cardíaca (sobretudo fosforilações do terminal amino) influem na regulação da função cardíaca, e subjazem à patogenia de certas doenças como a insuficiência cardíaca, a hipertrofia ventricular esquerda e a cardiomiopatia diabética.
De seguida expõem-se algumas dessas alterações pela sua importância clínica:
• Os resíduos de serina Ser-23 e Ser-24 do terminal N da troponina I cardíaca parecem ter importância na regulação da contractilidade cardíaca, e a sua fosforilação (mediada pela fosfocínase A) afecta a dinâmica e intensidade da frequência cardíaca (FC). Foi demonstrado, ainda, que essa fosforilação diminui a sensibilidade ao cálcio e aumenta a avidez daligação entre a TpI e o filamento fino.
• Os resíduos de serina Ser-42 e Ser-44 regulam a força das pontes cruzadas entre actina e miosina, tendo já sido demonstrado que a sua fosforilação (proteína cínase C) diminui a força de contracção muscular, através da depressão da cinética e tensão máxima dessas pontes.
• A fosforilação do resíduo de treonina Thr-143 aumenta a sensibilidade ao cálcio e diminui a cinética das pontes cruzadas.
• A fosforilação dos resíduos de serina Ser-43 e Ser-45, e da treonina Thr-144, conduz à inibição da contractilidade muscular, pela estabilização dos miofilamentos finos no seu modo de estado inactivo (11), contrariamente ao que se passa na cardiomiopatia hipertrófica, em que ocorre aumento da contractilidade muscular, por estabilização daqueles filamentos no modo de estado activo.
• Verificou-se que situações de isquemia / reperfusão moderadas induzem proteólise dos aminoácidos do terminal C da troponina I.
Finalmente, Genaro et al (12) apontaram, num trabalho recente, que a troponina I cardíaca (TpIc) é, certamente, a primadona dalusitropia (taxa de relaxamento muscular) induzida pela estimulação β-adrenérgica, podendo vir a constituir futuramente um alvo no tratamento específico da IC diastólica.
5 - DOSEAMENTO DA TROPONINA I
É possível distinguir a TpIc das suas isoformas esqueléticas, designadamente da lenta, pela singularidade da sequência de aminoácidos do terminal N (10).
Os testes de doseamento da troponina I actualmente existentes, usam anticorpos que se dirigem exclusivamente à isoforma cardíaca, pelo que se podem considerar testes específicos para a detecção de disfunção cardíaca.
A sensibilidade do teste é tanto melhor quanto maior for o número de anticorpos usados. De facto, num estudo de Estelle Le Moal et al (13), a combinação de anticorpos que mostrou maior sensibilidade clínica, consistiu no agrupamento de um anticorpo dirigido contra a porção central singular da troponina I, com dois anticorpos dirigidos contra epítopos do terminal N. Não obstante, existe uma grande diversidade de testes, nem sempre concordantes entre si, e com sensibilidades distintas, pela falta de padronização (ver adiante). Acresce, ainda, que na procura de maior sensibilidade se sacrifica a especificidade, podendo os testes ficar mais susceptíveis a interferências.
Seguidamente, faz-se uma breve revisão dos testes de doseamento, enfatizando aspectos relacionados com a sua padronização e cuidados a ter na interpretação dos resultados.
5.1 - Peculiaridades da molécula
Uma condição necessária ao uso de qualquer teste de diagnóstico, é a definição precisa do analito. Assim, antes de enveredar pela problemática dos testes de doseamento da troponina I importa esclarecer o que se pretende medir quando se aplicam esses testes, e o que se mede efectivamente.
Quando o analito é uma substância química bem definida (ex. colesterol, creatinina) torna-se fácil definilo. Porém, tratando-se de analitos complexos como é o caso dos biomarcadores proteicos, onde se inclui a TpIc, essa tarefa revela-se complicada, pela heterogeneidade molecular, seja intrínseca ou adquirida. Na realidade, a padronização dos testes é obviada, logo à partida, por este problema, tal como será analisado, à frente.
Cerca de 3 a 8% da TpIc encontra-se livre no sarcoplasma dos cardiócitos, mas a grande percentagem está incorporada nos miofilamentos finos. Após lesão miocárdica, a troponina TpIc é detectada na corrente sanguínea, sobretudo, na forma complexa de TpI-TpC (14).
Recentemente, Panteghini et al (15) propuseram que, para efeitos de doseamento, se definisse TpIc como sendo apenas a porção central da molécula da troponina I, uma vez que esta é a única região comum a todas as formas de troponina I, passíveis de serem encontradas em circulação (TpIc isolada, complexada com a TpC ou TpT, ou após certas modificações moleculares como a fosforilação ou oxidação). Segundo os mesmos autores dever-se-iam usar anticorpos dirigidos especificamente contra os epítopos dessa região da TpIc, permitindo homogeneização da reactividade dos testes, e portanto, padronização dos resultados. O problema é que, actualmente, os vários fabricantes continuam a usar anticorpos diferentes dirigidos contra diversos epítopos.
5.2 - Testes existentes no Mercado
A troponina C não tem utilidade na prática clínica porque o músculo cardíaco partilha a isoforma C da troponina com o músculo liso.
Efectivamente, apenas existem no mercado testes para determinação de duas troponinas: TpT e TpI. Somente um laboratório (Roche) comercializa o teste de determinação da troponina T. Contudo, actualmente, e tal como será abordado adiante, a troponina I é o biomarcador de eleição para diagnóstico do enfarte do miocárdio, sendo aquela que é mais frequentemente usada. Além disso, importa relembrar que a presente tese se centra na troponina I e no seu potencial valor prognóstico em doentes com agudização de DPOC. Deste modo, neste texto serão analisados, mormente, os testes de doseamento da troponina I, constando-se que existam pelo menos 18 testes diferentes no mercado (14, 16), divergindo entre si quanto ao tipo de reagentes usados, performance clínica, anticorpos monoclonais e tipo de epítopo da troponina reconhecido estes, bem como quais os valores cutoff estimados para o diagnóstico de enfarte agudo do miocárdio.
Há cerca de duas décadas atrás, quando surgiu o interesse pela troponina I, os primeiros testes baseavam-se em anticorpos policlonais dirigidos contra a troponina I, sendo bastante inespecíficos. Entretanto, começaramse a criar numerosos anticorpos monoclonais dirigidos contra aquelas regiões da molécula da troponina I, que se julgavam ser mais imunoreactivas. Seguidamente, usando várias combinações desses anticorpos, desenvolveram-se os primeiros testes de doseamento – Testes de 1ª geração. Estes testes caracterizavam-se por usar apenas um anticorpo, dirigido contra epítopos do terminal C ou N.
Apesar do entusiasmo inicial, depressa surgiu um problema relativamente aos testes de 1ª geração. De facto, a detecção da TpIc por imunoensaio pode ser afectada pelas diversas modificações (fosforilação, oxidação, degradação) que a molécula pode sofrer após ser libertada para a corrente sanguínea, pelo que, muitas vezes, o que se encontra em circulação, são pequenos fragmentos ou formas modificadas e não a TpIc original. Como os testes de 1ª geração apenas usavam um anticorpo dirigido contra os epítopos C ou N, que são sequências aminoacídicas susceptíveis de fosforilação ou oxidação (ver anteriormente), facilmente se depreende que, caso essas alterações moleculares tenham ocorrido, aqueles testes falham na detecção da TpI.
Assim, desenvolveram-se os testes de 2ª geração, que têm em conta, não só as formas livres de TpI, como também as formas complexadas, e as modificações póstranslacionais. São testes que usam conjuntos de dois ou três anticorpos, combinando um anticorpo especificamente dirigido contra a região central comum (ver acima) com um ou dois anticorpos dirigidos contra os epítopos dos terminais N e/ou C.
Pela corrente relevância clínica e laboratorial (17) destacam-se três testes de 2ª geração:
• Beckman Coulter Access Accu TnI:
Teste que usa 2 casas decimais na definição dos valores relativos aos limites de detecção (≤ 0,01 µg/L) e ao percentil 99 (0,04 µg/L, quer se use plasma ou soro).
Estudos recentes (18) mostraram ter acuidade semelhante à do Architect da Abbott, evidenciando sensibilidade de 85%, especificidade de 24%, e valores preditivos positivo e negativo de 10% e 95%, respectivamente, para morte nos primeiros 30 dias, e uma sensibilidade de 92%, especificidade de 25%, valor preditivo positivo de 9% e negativo de 98% para a ocorrência de enfarte do miocárdio aos 30 dias (usando o percentil 99 como valor cutoff). O valor para o qual se verificou um coeficiente de variação (CV) = 10% ficou estabelecido pelos 0,014 µg/L.
• Abbott Diagnostics Architect STAT Troponin-I:
Este teste era bilaminado, mas recentemente foi melhorado (19), através da adição de um novo anticorpo, sendo actualmente trillaminado (tal como o TnI-Ultra ADVIA Centaur, da Siemens). Assim, usa dois anticorpos dirigidos contra epítopos na região específica da forma cardíaca da troponina I (aminoácidos 27 – 40 e 41 – 49), e um anticorpo dirigido contra o epítopo 87 – 90. Actualmente, graças aos aperfeiçoamentos de que tem sido alvo, é considerado um teste altamente sensível para determinação da troponina I.
Usa 3 casas decimais, com limites de detecção que variam (17, 20) entre valores ≤ 0,009 e ≤ 0,010 µg/L. Contrariamente aoAccu TnI, os percentis 99 no plasma e no soro, correspondem a valores de troponina I distintos, respectivamente 0,012 µg/L e 0,025 µg/L.
Todavia, com a versão mais recente, estas diferenças não são estatisticamente significativas (21).
• ADVIA / Centaur CP TnI-Ultra, Siemens Medical Solutions Diagnostics:
É um método de quimioluminescência, trilaminado, porque usa dois anticorpos monoclonais dirigidos contra epítopos existentes nos aminoácidos 41 – 49 e 87 – 91, bem como um marcador anticorpo policlonal intestinal (marcado com Ester de Acridínio) dirigido contra as sequências 27 – 40. Também usa três casas decimais, e o limite de detecção situa-se nos 0,006 µg/L, mostrando um coeficiente de variação de 10% para valores de TpIc = 0,03 ng/ml.
Num estudo de Fred et al. (22), este teste mostrou sensibilidade de 94% e especificidade de 84% para diagnóstico de enfarte agudo do miocárdio, nas primeiras 6h a 24h, para um percentil 99 ≤ 0,04 µg/L. Não obstante, a literatura é contraditória nos estudos de comparabilidade da acuidade diagnóstica. Casals et al. (23) alegaram ter acuidade semelhante á do Accu TnI, mas recentemente, Venge et al (17) mostraram que este último tem maior sensibilidade diagnóstica.
Como nota final importa referir que o TnI-Ultra não é afectado pelas diluições da troponina I com plasma rico em triglicerídeos ou proveniente de doentes com artrite reumatóide (24). Mas parece ser influenciado por anticorpos heterofílicos (25).
Com os testes de 2ª geração,identificaram-se pessoas com valores detectáveis de TpIc, ainda que inferiores ao percentil99 da população de referência. E foidemonstrado que esses níveis baixos de troponina I em circulação, tinham implicações prognósticas.
Posto isto, os investigadores questionaram-se, quão baixo poderiam descer até desaparecer o valor preditivo da troponina I?
No sentido de esclarecer esta hipótese, foram-se aperfeiçoando os testes de doseamento da troponina, criando-se testes ultrasensíveis, como o Erenna Immunoassay System (26), previamente designado Zeptx System. Neste sistema, a técnica de imunoensaio por fluxo está acoplada a um instrumento digital de contagem de moléculas. John et al. (26) testaram este sistema em indivíduos saudáveis tendo mostrado limites de detecção para valores de 0,2 ng/L com coeficiente de variação de 10% para valores entre 0,78 e 1,6 ng/L. As vantagens (27) deste tipo de teste incluem a necessidade de menor quantidade de amostra, e um tempo de incubação mais curto. Todavia, mostrou sensibilidade inferior quando comparado com os sistemas convencionais.
Recentemente, Reichlin e colegas (28) testaram a acuidade diagnóstica de quatro testes ultrasensíveis (dois para troponina I e dois para a troponina T) em comparação com o ADVIA Centaur TnI-Ultra, e verificaram que a acuidade diagnóstica daqueles quatro era significativamente superior à deste último, com um valor preditivo negativo entre 97 – 99%. Importa referir, a título de curiosidade, que esses quatro testes ultrasensíveis foram: 1 – Abbot-Architect TnI (já referido acima), com limite de detecção de 0,01 ng/ml, um percentil 99 de 0,028 ng/ml, e um CV < 10% para valores de 0,032 ng/ml; 2 – Elecsys 2010 system da Roche TnT (teste de 4ª geração) com limite de detecção de 0,01 ng/ml, percentil 99 de 0,01 ng/ml, e um CV < 10% para valores de 0,035 ng/ml; 3 – Elecsys 2010 system da Roche High Sensitive TnT com limite de detecção de 0,002 ng/ml, percentil 99 de 0,014 ng/ml e um CV < 10% para valores de 0,013 ng/ml; e 4 – Elecsys 2010 system da Roche TnI com limite de detecção de 0,10 ng/ml, percentil 99 de 0,16 ng/ml e CV < 10% para valores de 0,30 ng/ml.
5.3 - O problema da padronização dos testes
Tal como mencionado anteriormente, a troponina é o principal biomarcador utilizado no diagnóstico de enfarte agudo do miocárdio. Convém, portanto, que os testes em comercialização sejam padronizados. Contrariamente ao que se passa com a TpT, cujos testes são comercializados apenas por um fabricante (Roche), no caso da troponina I existem diversas companhias a comercializar diferentes testes de doseamento, e na literatura (15) existem relatos de coeficientes de variação superiores a 10% quando confrontados resultados dos diferentes laboratórios, o que constitui um entrave à investigação científica, pela impossibilidade de fazer comparações.
A falta de padronização dos testes de TpI é uma realidade porque cada fabricante usa materiais “padrão” diferentes e anticorpos dirigidos contra epítopos distintos. Alguns autores (29) recomendam o uso de materiais padrão reconhecidos internacionalmente ou pelo menos, materiais de referência universal, tendo em conta determinantes específicos da molécula da troponina I, bem como sistemas de medição padrão. A introdução do sistema ADV, em 2005, nos analisadores AxSym e Architect da Abbott, permitiram uma redução dos CV para 8,2% e 4,3%, respectivamente.
Condizente com esta problemática, recentemente foram publicados (15) alguns preceitos a cumprir quando da padronização dos testes de doseamento da troponina I. Esse documento dá ênfase ao sistema de medição de referência, que dizem envolver três componentes: procedimento de referência, definição padrão do analito e materiais de referência para calibração.
O procedimento de referência consiste na acção de certificar o valor dum material de referência secundário (analito presente numa matriz complexa (ex. soro dos doentes com enfarte agudo do miocárdio, que dispõem de valores elevados de TpI) comparável àquela existente na amostra a dosear), calibrado através do uso de um material de referência primário. Actualmente, o procedimento de referência universalmente advogado faz-se por imunoensaio, usando anticorpos monoclonais específicos contra os epítopos centrais da molécula da TpI, apesar de, como qualquer procedimento de medição indirecta, ser muito técnico-dependente. Isto porque, os métodos não imunoquímicos (como a espectrometria de massa) que seriam de preferir para efeitos de padronização, ainda não têm sensibilidade suficiente para a medição directa da TpI, sobretudo naquelas situações em que aquela se encontre em níveis muito baixos.
A ambiguidade na definição do analito TpI, já explorada, anteriormente, neste texto, obsta a padronização. Todavia, tem-se vindo a fazer um esforço no sentido de promover o conceito da TpI interessar apenas a zona central da sua molécula (15), por ser a única estável do ponto de vista estereoquímico.
Katrukha et al. (30) sugeriram que o material de referência primária, para o calibrador, deveria ser constituído por concentrações equimolares de troponinas I, T e C, de modo a representar a maior e mais natural forma antigénica de troponina encontrada no sangue – o complexo troponínico. O SRM 2921 (desenvolvido por American Association for Clinical Chemistry, National Institute of Standards and Technology e o IFCC) aspirou a material de referência secundária universal (MRSU). Porém, verificou-se que a suaintrodução no mercado não melhorava a comparabilidade dos valores de troponina I sérica, de amostras humanas, fazendo suspeitar que o uso de um MRSU, só por si, não chega para padronizar os testes. De facto, apesar do SRM 2921 pretender mimetizar a principal forma molecular da TpI encontrada nas amostras biológicas, os analitos presentes no material de referência e aqueles presentes na amostra, são, definitivamente diferentes.Acresce, ainda, que apesar da remoção da matriz estrutural, o processo de purificação pode conduzir a modificações parciais da molécula da troponina, afectando a reacção imunológica (pelo menos em alguns testes). Além disso, verificou-se que o SRM 2921 não tem estabilidade ao longo do tempo, pelo que, actualmente, se considera que o melhor material e referência secundária (31) é uma amostra de sangue oriunda de doentes com elevação da TpI.
Além do problema da padronização dos testes de determinação da troponina I, existe o problema da ambiguidade na definição dos valores cutoff considerados no diagnóstico de enfarte agudo do miocárdio. De facto, enquanto alguns laboratórios usam o valor associado a um coeficiente de variação inferior a 10%, outros usam o valor equivalente ao percentil 99 duma população de referência.
Em 2008 foi publicado (32) o resultado duma auditoria efectuada no âmbito do estudo piloto CARMAGUE, onde consta que apesar da maioria dos laboratórios usar a troponina (T ou I) como principal biomarcador de necrose miocárdica, não detêm frequentemente protocolos de actuação, e os limites de decisão utilizados são bastante variáveis (em 39% dos casos usavam o valor com cv 10% e em 35% dos casos, o percentil 99; os restantes 26% usavam outros valores cutoff perfeitamente arbitrários). Não há razão para que isto se continue a verificar, uma vez que estudos recentes (33) corroboraram a hipótese que o percentil 99 deve ser o único valor cutoff usado. No entanto, nada obsta a que o valor absoluto deste percentil seja diferente conforme a população estudada.
5.4 - Interferência com doseamento
Reconhecem-se, hoje, vários parâmetros que podem influenciar os resultados laboratoriais quando se trata do doseamento da TpI. Por uma questão didáctica, far-se-á uma exposição em separado, conforme se tratem de factores pré-analíticos, analíticos ou pós-analíticos.
Os erros pré-analíticos abrangem desde a simples identificação errada dos tubos de colheita, com subsequente troca de amostras, e o manuseio inadequado do sangue, até situações mais complexas como aquelas relacionadas com o fabrico do próprio teste de doseamento. Erros envolvendo o procedimento de diluição da TpI, encaixam neste último grupo. Tze-Kiong e colegas (34) defenderam que a diluição com soro fisiológico ou água bi-destilada (usados por vários fabricantes) pode conduzir à leitura errada dos valores plasmáticos da troponina I, sendo de preferir o uso de um diluente específico, diluente A da Access, por ter sido o único que demonstrou ter, simultaneamente, menor risco de ligação inespecífica dos anticorpos, e o meio iónico mais adequado e com pH ideal para a ocorrência da interacção anticorpo-antigénio troponínico.
A reacção anticorpo – antigénio troponínico pode ser afectada por vários factores como a presença de lípidos na amostra, ou de certos fármacos / drogas, e a ocorrência de hemólise.
Na literatura, foi também descrito que a presença de vestígios de fibrina nas amostras de sangue, podia conduzir a resultados falsos positivos, e, recentemente, Carine e colegas (35) propuseram um procedimento com intuito de prevenir este tipo de erro, através da ultracentrifugação (6700xg durante 5 minutos) das amostras.
Nos últimos anos têm-se vindo a fazer várias alterações nos tubos de colheita de amostras biológicas, designadamente substituição do vidro por plástico, e a introdução de gel polimérico e activador de coágulos. Alguns autores (36) sugeriram que o doseamento da troponina I poderia ser afectado pelo material dos tubos de colheita (particularmente pela heparina e gel separador). Não obstante, foi demonstrado que a troponina I, contrariamente à mioglobina e CK-MB, não éinfluenciada por estes factores.
A procura de testes cada vez mais sensíveis, através da incorporação de maior nº de anticorpos, condicionou um maior potencial para a interferência com anticorpos heterofílicos, presentes no soro humano. Foidemonstrado que estes se podem ligar quer aos anticorpos de captura (aqueles que reagem em primeiro lugar com a TpIc) quer aos de marcação (aqueles que vão marcar os complexos anticorpo-TpIc previamente formados), mimetizando a troponina I, através da formação de complexos anticorpoantigénio artefactuais, e produzindo resultados errados. Este mecanismo de interferência analítica foi descrito, pela primeira vez na década de 70 por Prince e colegas (37), a propósito de testes serológicos da hepatite B, e no ano 2000, Yeo e colegas (38) alertaram para a sua ocorrência também em testes de doseamento da TpI, concretamente, testes de 2ª geração.
A maioria dos testes comercializados em 2008 era bifaseada, isto é, processava-se em 2 etapas, usando dois anticorpos dirigidos contra dois epítopos diferentes da troponina I. Yussheng Zhu e colegas (25) relataram, em 2008, numa carta ao editor, o primeiro caso de interferência de um teste ultra-sensível (ADVIA Centaur System da Siemens, denominado Ultra-TnI) por anticorpos heterofílicos, num doente admitido por pneumonia de aspiração. De facto, os imunoensaios ditos de ultrasensíveis são trifaseados (incorporando 3 anticorpos diferentes dirigidos contra certos epítopos da troponina I), e parecem ser cerca de duas vezes (25) mais propensos à interacção com anticorpos heterofílicos que os seus congéneres bilaminados.
Ainda relativamente à usurpação dos resultados dos testes da TpI, importa fazer referência aos doentes em programa regular de hemodiálise, pela potencial interferência da membrana do dialisador na depuração e consequente doseamento da troponina I, em circulação. Giuseppe e colegas (39), num estudo caso-controlo, comparando valores de TpIc antes e após sessão de hemodiálise, verificaram que havia uma descida estatisticamente significativa, apenas nos casos em que se usaram membranas de alto fluxo.
5.5 -Consequências do uso de testes ultrasensíveis
As primeiras horas após o início dos sintomas de enfarte do miocárdio, são cruciais, pela possibilidade de intervenção terapêutica precoce, interessando, por isso, usar marcadores passíveis de serem detectados em concentrações mínimas, o mais cedo possível. Porém, com os testes contemporâneos, de 2ª e 3ª geração, os biomarcadores de subida precoce (mioglobina,isoformas da CK-MB), clinicamente sensíveis mas não específicos, deixaram de ter utilidade diagnóstica (20), uma vez que, mesmo nestas circunstâncias, a troponina I mostrou maior sensibilidade e especificidade.
Com a introdução de testes altamente sensíveis no mercado, temeu-se um incremento no número de testes solicitados aos laboratórios, e a submissão abusiva de indivíduos saudáveis a terapêuticasinvasivas. Conquanto, recentemente, num estudo de Melanson et al. (40), constatou-se um aumento do número de testes positivos, como seria de esperar, sem acréscimo da quantidade de testes solicitados.
Acresce ainda, que a par do desenvolvimento de testes altamente sensíveis, os critérios de diagnóstico de enfarte agudo do miocárdio foram actualizados (ver abaixo) e estudos recentes mostraram que estes novos critérios são factores preditivos significativos de mortalidade aos 3 anos (41). Deste modo, o acréscimo no nº de enfartes diagnosticados não é de todo supérfluo.
Com limites de detecção cada vez mais baixos, os investigadores e clínicos deparam-se, actualmente, com outro problema – variabilidade inter e intra-individual, bem como ligações inespecíficas de baixo nível – factores até agora descurados, por ocorrem abaixo do limite de detecção dos testes. Ulteriormente, Wu et al. (42) mostraram que a variabilidade intra-individual (VIntra) em indivíduos saudáveis é pequena quando comparada com a inter-individual (VInter), e o índice de individualidade (VIntra/VInter) encontrado foimínimo, sugerindo que para interpretação de valores tão baixos de TpI, é mais útil fazer doseamentos seriados e monitorizar as alterações da TpI em cada indivíduo, do que compará-los com os valores duma população de referência.
5.6 - Breve referência aos testes rápidos
Ultimamente têm sido descritos alguns testes rápidos para doseamento da troponina I, úteis como primeiro teste de diagnóstico, em contexto de emergência pré-hospitalar. A título de exemplo, descrevem-se os seguintes:
6 -DETECÇÃO DA TROPONINA I E SIGNIFICADO CLÍNICO• Pathfast: Sistema de imunoensaio enzimático por quimioluminescência altamente sensível, que usa cartuchos descartáveis de reagente, para utilização única, permitindo medição da TpI, em contexto de emergência pré-hospitalar. Usa material de referência padrão, o SRM 2921. É um sistema fácil de usar, rápido e sensível, tendo-se constatado (43) que a sua performance analítica é semelhante à dos testes laboratoriais convencionais.
• Biosensor EOC: Sistema que combina a quimioluminescência por ELISA com um detector de imagem equipado com uma câmara CCD, para detecção de sinal quimioluminométrico, mostrou ter alta sensibilidade, quando comparado com os testes rápidos convencionais (44). • I-STAT point-of-care troponin I test, Abbott: teste rápido recentemente introduzido em alguns hospitais (em contexto de emergência) que mostrou elevado coeficiente de correlação (r=0,97) com o teste convencional (ARCHITECT STAT troponin-I), em termos de resultados positivos, mas exibiu menor sensibilidade, sobretudo para pequenas elevações da troponina I, nas primeiras horas após enfarte (45).
6.1- Libertação, degradação e depuração da troponina I
A libertação da troponina cardíaca para a corrente sanguínea pode dar-se quer após lesão irreversível dos miocardiócitos (necrose miocárdica) quer após dano reversível (lesão cardíaca isquémica como na angina instável ou outro tipo de lesão reversível) por alterações na permeabilidade da membrana celular.
No caso do enfarte do miocárdio, inicialmente (nas primeiras 4h – 6h) ocorre a libertação de uma pequena quantidade de troponina, que provém do reservatório citosólico (ver atrás), verificando-se, depois, um pico por volta das 48h – 72h, correspondendo à troponina do reservatório estrutural, que começa a ser libertada para o plasma graças à destruição das miofibrilas, quando da necrose dos miocardiócitos. Seguidamente observa-se uma fase caracterizada pela presença de valores elevados de troponina, ainda que em evolução decrescente, que pode persistir por 5 – 10 dias (16, 46).
Noutras situações que não o enfarte do miocárdio, pode suceder uma das seguintes alterações: uma subida e descida rápidas (em 24h – 48h) da troponina plasmática (em casos de isquemia do miocárdio, sem enfarte), com um pico tendencialmente inferior ao que acontece no caso de enfarte, ou então a presença de valores baixos de troponinemia, razoavelmente constantes ao longo de algum tempo (doenças cardíacas não isquémicas e patologias não cardíacas, como especificado abaixo), reflectindo lesão reversível do miocardiócito, somente com libertação da troponina citosólica.
Tal como explorado anteriormente neste texto, a maior parte da troponina libertada para o plasma encontra-se sob a forma de complexos moleculares (maioritariamente TpI-TpC, mas também TpI-TpC-TpT) ou então como produtos resultantes da sua degradação intra ou extracelular (16), sendo libertada na forma livre e intacta apenas numa pequena percentagem (cerca de 3-10%).
Relativamente à depuração da troponina sabe-se pouco (16), antevendo-se, contudo, que possa ser eliminada mormente pelo sistema reticuloendotelial, atendendo às grandes dimensões da molécula/complexos moleculares. Conquanto, os produtos de degradação da troponina são passíveis de serem excretados por via renal, embora ainda não tenha sido confirmado por estudos clínicos.
6.1.1 Significado clínico de valores detectáveis de troponina I
Já na década de 1990 se vislumbrou que a troponina I, apesar de ser um marcador específico de mionecrose cardíaca, poderia ser detectada em níveis variáveis noutras situações que não os síndromes coronários agudos (47), designadamente em doenças pulmonares, na sépsis, em doenças cardíacas não isquémicas como a cardiomiopatia dilatada e a insuficiência cardíaca não isquémica, nainsuficiência renal, nainfecção pelo VIH (vírus da imunodeficiência humana), no hipotiroidismo, lúpus eritematoso sistémico e em certas doenças musculares e do sistema nervoso central. Nessa altura especularam quanto à etiologia desta troponinemia, presumindo que resultasse de necrose miocárdica à escala das miofibrilas, sem que ocorresse enfarte do miocárdio.
Desde então, vários autores (48, 49, 50) têm vindo a investigar a etiologia da troponina I detectada em doentes com coronárias angiograficamente normais, tendo-se descortinado, um pouco melhor, a vastidão de patologias e mecanismos patofisiológicos potencialmente conducentes à lesão miocárdica que subjaz à libertação de pequenas quantidades de TpI para o plasma.
Por uma questão didáctica e servindo-me da estruturação efectuada por Mahajan et al. (49), podemos organizar as causas de troponinemia, na ausência de síndromes coronários agudos (ou mais correctamente, na ausência de doença coronária angiograficamente significativa), em cinco grandes grupos, conforme especificado abaixo:
1. Lesão directa dos miocardiócitos:
Pode ocorrer lesão directa dos miocardiócitos em situações de inflamação do miocárdio (51) (sobretudo na miocardite, mas também pericardite), estimulação eléctrica excessiva do coração (electrocussão, descargas do CDI (cardiodesfibrilador implantável), desfibrilação ou cardioversão eléctrica, ablação auriculoventricular), lesão mecânica directa (como a que acontece no contexto de cirurgia valvular ou cirurgia de revascularização coronária, ou em caso de contusão do miocárdio) ou agressão química, como acontece com o uso de quimioterapia cardiotóxica (particularmente das antraciclinas).
2. Diminuição no fornecimento de oxigénio:
O espasmo das artérias coronárias é uma causa importante de troponinemia, potencialmente fatal (morte súbita cardíaca), que cursa com isquemia transitória dos miocardiócitos por vasoconstrição secundária à hiper-estimulação α-adrenérgica (49, 50,52). Uma outra causa (menos frequente) de vasoconstrição coronária transitória relaciona-se com uma variação anatómica da descendente anterior, que faz parte do seu trajecto dentro do miocárdio, e, portanto, é comprimida por este durante a sístole.
As situações de choque (49), quer pela hipotensão arterialquer pela taquicardia, condicionam diminuição da perfusão coronária, e o seu tratamento, usando vasopressores, pode contribuir, também, para a lesão dos miocardiócitos, pela hiper-estimulação α-adrenérgica (ver acima).
A anemia, por diminuição do aporte de oxigénio ao tecido cardíaco, pode causar mionecrose e originar troponinemia, sobretudo nos idosos. As hemorragias, pela diminuição do volume de sangue circulante e pela anemia, também comprometem o fornecimento de oxigénio e nutrientes ao miocárdio.
Os estados de hipercoagulabilidade (como nas doenças auto-imunes e neoplásicas) podem cursar com micro-oclusão vascular e micro-enfartes. De igual modo, em casos de dissecção das coronárias ou da artéria aorta, o fluxo coronário fica comprometido, podendo resultar em lesão miocárdica hipóxica.
3. Aumento das necessidades de oxigénio:
A hipertrofia ventricular esquerda, por aumento da massa miocárdica, implica um aumento no consumo de oxigénio e nutrientes, podendo ocasionar lesão miocárdica caso essas necessidades não sejam satisfeitas.
A taquicardia, supra-ventricular (53) ou ventricular, pode lesar os miocardiócitos pelo efeito conjunto do aumento do consumo de O2 aliado à diminuição da perfusão coronária pela redução do tempo de diástole (49,52,54).
Vários autores (49,52,54,55) verificaram níveis detectáveis de troponina I em doentes com insuficiência cardíaca (IC) crónica estável, reflectindo, provavelmente, uma perda progressiva de miocardiócitos viáveis, por isquemia/necrose subendocárdica ou apoptoseinduzida pelo estiramento miocárdico (56, 57, 58) (definido como a alteração percentual da estrutura do miocárdio após aplicação de tensão, relativamente ao seu comprimento inicial (59)) ou por citocinas tóxicas (56), elevadas no contexto da IC. A tensão sobre a parede ventricular, resultante de pressões tele-diastólicas elevadas, parece induzir a entrada de cálcio nos miocardiócitos, com activação subsequente das µ-calpaínas, produzindo proteólise da troponina I (60), independentemente da ocorrência ou não de isquemia, com libertação dos seus fragmentos para o plasma. Todavia, também já foi demonstrado, experimentalmente, que os miocardiócitos respondem ao estiramento através das integrinas (receptores transmembranares glicoproteicos), com libertação da troponina I na formaintacta (61). Um outro mecanismo que pode contribuir para o aumento da troponina plasmática em doentes com IC é a estimulação simpática e do sistema renina-angiotensina (50).
No caso da embolia pulmonar aguda, qualquer que seja a extensão, o aumento súbito de pressão ao nível do ventrículo direito, implica um aumento marcado no consumo de O2 e nutrientes, podendo causar lesão miocárdica nesse contexto (49), agravada pela hipoxia resultante do desequilíbrio entre a ventilação e a perfusão. Além disso, é provável que a isquemia subendocárdica e a tensão mecânicaintramuralcausadas pelo aumento da pós-carga ao nível do ventrículo direito (62), contribuam, também, para a libertação da troponina cardíaca para o plasma.
Outra entidade que se pode acompanhar de troponinemia é a hipertensão arterial (49,50,56), sobretudo se associada a hipertrofia ventricular esquerda e proteinúria, reflectindo, provavelmente, lesão miocárdica subsequente ao estiramento da parede ventricular. Do mesmo modo, a detecção de troponina I plasmática pós pericardiocentese descrita por Nunes et al. (63) pode dever-se ao estiramento da parede ventricular, subsequente ao aumento súbito do raio ventricular e, de acordo com a lei de Laplace, da tensão sobre a parede ventricular, pós drenagem do líquido pericárdico.
A doença cardíaca valvular, particularmente da válvula aórtica (estenose ou regurgitação), implica um aumento no consumo de O2 pelo miocárdio (maior tensão na parede), agravado por uma limitação importante do fluxo coronário, com lesão miocárdica subsequente.
As situações de hiper-estimulação simpática (hemorragia subaracnoideia, acidente vascular cerebral, feocromocitoma, consumo de drogas simpaticomiméticas como a cocaína), por desequilíbrio ao nível do sistema nervoso autónomo (49,50), causam lesão miocárdica por vasoconstrição α-adrenérgica.
4. Diminuição do fornecimento de oxigénio a par de um aumento na sua necessidade:
A sépsis (16,49,50,56) é o paradigma da condição que conjuga um aumento do consumo de O2 por parte do miocárdio (pelo estado inflamatório agudo, por exemplo) com uma diminuição no seu fornecimento (pelas alterações hemodinâmicas associadas, pela hipoxia e pela formação de trombos microvasculares decorrentes do estado pró-trombótico). Pensa-se que a lesão miocárdica neste caso possa resultar também de lesão directa pelas toxinas bacterianas (particularmente do Streptococcus pneumoniae (16)) e pelas citocinas inflamatórias. As taquiarritmias (49,50), referidas acima, também obrigam a um aumento do consumo de O2 por parte do miocárdio, ao mesmo tempo que diminuem a sua perfusão, pelo inerente encurtamento da diástole, principal fase do ciclo cardíaco em que ocorre perfusão do miocárdio. A hipertrofia ventricular esquerda por aumento da massa miocárdica obriga a um aumento no consumo de O2, comprometendo, além disso, a perfusão miocárdica através da redução da reserva do fluxo coronário, subsequente à necessária remodelação coronária, e conduzindo àisquemia subendocárdica, e à libertação da troponina (50).
O exercício físico extremo ou de resistência (16,50) pode levar a aumentos significativos da troponina plasmática, não só pelo aumento do consumo do O2 por parte do miocárdio, como também por uma potencialdiminuição da perfusão coronária por vasospasmo induzido pelas catecolaminas endógenas libertadas quando do exercício.
5. Outras causas:
Nas doenças infiltrativas do miocárdio (como a amiloidose e a sarcoidose) pode ocorrer lesão da célula muscular cardíaca por compressão directa (graças à acumulação extracelular de material), estimulando a libertação da Tp (16,49,50).
A cetoacidose diabética, também pode lesar os miocardiócitos, por agressão metabólica (metabolismo anaeróbio) directa.
Os níveis mais elevados de Tp em doentes com insuficiência renal crónica (49,50,56) podem dever-se a uma diminuição na sua excreção (pensa-se que o principal mecanismo de eliminação da troponina seja pelo sistema reticuloendotelial, dadas as grandes dimensões da molécula; contudojá foidescrito que a troponina T se pode fragmentar em pequenas moléculas passíveis de serem eliminadas via renal). Conquanto, o aumento da massa ventricular esquerda e a ocorrência de pequenas áreas de necrose miocárdica à escala das miofibrilas, podem contribuir também para essas elevações da troponina. Uma outra explicação possível é a ocorrência de “inflamação” do músculo cardíaco por agressão directa das toxinas urémicas (16,51).
Alguns autores (64) encontraram, ainda, níveis elevados de troponina I em certos doentes com cirrose hepática, mostrando uma associação significativa com a diminuição do índice do volume de ejecção e da massa ventricular esquerda, mas não com a gravidade da cirrose. Outros autores, estudaram doentes com rabdomiólise, e verificaram que a troponina cardíaca se encontra muitas vezes elevada nesses doentes, mas que esse aumento não se correlaciona com a lesão muscular, insuficiência renal ou com factores de risco cardiovascular, parecendo mais provável que resulte da patologia conducente à rabdomiólise, designadamente a hipotensão arterial, o uso de drogas ilícitas e a sépsis (51).
Por último, mas não menos importante, não se pode desprezar uma potencial subida falsa da troponina cardíaca, pela interacção dos reagentes do teste com anticorpos heterofílicos, tal como exposto anteriormente, tendo sido já descrita em doentes com artrite reumatóide (49,54) e exacerbação aguda de DPOC por infecção respiratória (65). Acresce, ainda, a possibilidade de ocorrerem resultados falsamente positivos por interferência com a bilirrubina, e em casos de hemólise (16).
Após esta pequena exposição percebe-se a diversidade de causas e mecanismos fisiopatológicos subjacentes a pequenas subidas de troponina cardíaca plasmática. Numa tentativa de uniformização, recentemente, houve (66) quem sugerisse a criação de uma nova entidade nosológica – Troponinemia ligeira de origem indeterminada (TLOI) – onde se deveriam incluir todos aqueles doentes que apesar de apresentarem níveis detectáveis de Tp, não cumprissem os critérios de diagnóstico de enfarte do miocárdio.
Todavia, a nova definição de enfarte agudo do miocárdio (EAM) trouxe, seguramente, maior dificuldade na procura de uma interpretação para pequenas elevações da troponina cardíaca, nomeadamente no que se refere ao chamado enfarte do miocárdio tipo II (ver abaixo). De facto, muitas doenças em que se têm vindo a detectar pequenas subidas plasmáticas de Tp, como ainsuficiência cardíaca, as exacerbações da DPOC, a sépsis, o tromboembolismo pulmonar, a fibrilação auricular, etc, cursam com hipotensão arterial e/ou hipoxemia, os mecanismos fisiopatológicos do enfarte do miocárdio tipo II.
Efectivamente, no momento actual, embora aclarada a etiologia de pequenas elevações de troponina plasmática, a sua abordagem na prática clínica continua polémica. De facto, Blich et al. verificaram que a grandeza do aumento da troponina nestas circunstâncias não é discriminativa (52), não se podendo definir com precisão quem tem ou não síndrome coronário agudo (área abaixo da curva ROC =0,63). De qualquer forma, alguns estudos mostraram que doentes com níveis detectáveis de troponina I plasmática, sem síndrome coronário agudo, têm pior prognóstico que os doentes com enfarte agudo do miocárdio (52), provavelmente, por carência terapêutica atempada e eficaz, dirigida às potenciais patologias subjacentes, tal como descrito acima. É, portanto, crucial identificar sempre que possível essas causas subjacentes.
Um novo conceito introduzido recentemente é o da estratificação do risco cardiovascular em doentes com subidas mínimas de troponina plasmática (abaixo do percentil 99), parecendo haver particular interesse no uso da CK-MB e da PCR (67).
Finalmente, é importante referir que, contrariamente ao que se passa num contexto de síndrome coronário agudo, não existem ainda ensaios clínicos randomizados e controlados que permitam avaliar a pertinência da instituição de terapêuticas (como antiagregantes plaquetários, estatinas, IECAs e bloqueadores β) visando redução do risco cardiovascular traduzido por estas pequenas elevações plasmáticas da troponina cardíaca (50).
6.2 -Uso da troponina I como exame auxiliar de diagnóstico
É do conhecimento actual (68,69) que o mecanismo subjacente aos síndromes coronários agudos (SCA) envolve ruptura e/ou erosão de uma ou várias placas ateroscleróticas, conduzindo ou não à necrose dos miocardiócitos. De igual forma, sabe-se que o evento final da necrose miocárdica/disfunção cardíaca passa por uma série de fases – formação da placa aterosclerótica —› ruptura / erosão da placa —› trombose arterial —› isquemia —› necrose —› remodelação / reparação do miocárdio – para as quais existem hoje marcadores específicos.
Clinicamente interessariam os biomarcadores de isquemia (68) (ex. ischemia modified albumin, unbound free fatty acids, choline), uma vez que permitiriam tomar, oportunamente, medidas de prevenção do enfarte do miocárdio. Mas, problemas relacionados com a instabilidade das moléculas, a imperfeição de alguns testes e os custos elevados de outros, tornam impraticável, por enquanto, o seu uso na prática clínica (69).
Assim, os biomarcadores presentemente usados no diagnóstico de doença cardíaca isquémica são marcadores de necrose miocárdica, e relativamente a estes importa que identifiquem o mais rapidamente possível o enfarte agudo do miocárdio para a implementação eficaz duma medicina baseada na evidência.
A troponina cardíaca consiste num marcador específico de lesão dos miocardiócitos e, graças aos testes de doseamento ultrasensíveis actualmente disponíveis, dispõe de uma razão sinal/“ruído de fundo” muito alta. Acresce ainda o facto de ter mostrado elevada sensibilidade e especificidade no diagnóstico de necrose miocárdica. Graças a estas características, a troponina cardíaca (I ou T) tornou-se no biomarcador padrão para o diagnóstico do enfarte do miocárdio (68-70), preterindo a Ck-MB, e mesmo novos marcadores precoces de necrose miocárdica, como o H-FABP (heart-type fatty acid binding protein) (71).
No caso do enfarte agudo do miocárdio com supradesnivelamento do segmento ST (EMCSST), a troponina serve apenas para confirmar o diagnóstico inicial (porque na prática clínica as alterações electrocardiográficas não têm especificidade de 100%) (72). De resto é usada, maioritariamente, como factor de prognóstico (73,74) (extensão do enfarte), tal como será desenvolvido na próxima secção.
Caso não se observe elevação do segmento ST e os doentes apresentem baixo risco de doença coronária, a troponina é útil sobretudo como marcador de exclusão, isto é, se vier negativa nas 12 – 14h seguintes ao início dos sintomas é pouco provável que se trate dum EAM (68). De qualquer forma, o facto de ser negativa não excluia possibilidade duma estenose coronária hemodinamicamente significativa, pelo que é forçoso fazer prova de esforço convencional ou cintigrafia de perfusão do miocárdio. Se, porém, a troponina for positiva, faz-se o diagnóstico de EAM (ver abaixo), identificando doentes que beneficiarão de medidas como o uso de heparina de baixo peso molecular, inibidores da glicoproteína IIb/IIIa e/ou revascularização coronária.
Quando, na ausência de alterações electrocardiográficas (ECG) sugestivas de isquemia (alterações do ST –T ou bloqueio completo de ramo esquerdo de novo), a probabilidade pré-teste para enfarte do miocárdio é baixa, a troponina é amplamente útil como factor de exclusão se vier negativa após duas determinações (à admissão e 6 – 9h após). Se vier positiva, importa, sobretudo, analisar a curva dinâmica e a percentagem de variação entre os diversos doseamentos, antes de fazer o diagnóstico de EAM (68).
Em 2007 foi publicada uma actualização dos critérios de diagnóstico de enfarte agudo do miocárdio (75) que refere o percentil 99 do limite superior do valor de referência, preferencialmente, da troponina como condição necessária para o diagnóstico, mas não suficiente, uma vez que obriga à existência de pelo menos outro critério (sintomas deisquemia cardíaca, alterações ECG sugestivas deisquemia, desenvolvimento de ondas Q patológicas no ECG, ou evidência imagiológica de novo de perda de miocárdio viável ou alterações da motilidade da parede). Um novo conceito trazido por esta redefinição é o da classificação do EAM mediante a causa da isquemia. De facto, a necrose miocárdica nem sempre resulta de isquemia decorrente da ruptura de placas ateroscleróticas e oclusão coronária subsequente (EAM tipo I), podendo advir dum desequilíbrio entre o fornecimento / consumo de O2 e nutrientes (anemia, hipo ou hipertensão arterial, arritmia, vasoconstrição ou espasmo arterial), o chamado EAM tipo II. Além disso, uma subida da Tp (se 3x o limite superior do valor de referência) apósintervenção coronária percutânea é, presentemente, diagnóstica do EAM tipo IV, e após cirurgia de revascularização coronária (se subida de 5x acima do limite superior do valor de referência), do EAM tipo V.
O EAM tipo III distingue-se dos outros tipos por não depender da troponina cardíaca. De facto, designa-se assim quando o indivíduo morre subitamente antes de ter sido possível colher sangue para doseamento dos marcadores bioquímicos de lesão miocárdica ou de ter decorrido o tempo necessário para que fossem detectados no plasma, mas em que previamente se tenham documentado sintomas isquémicos ou alterações electrocardiográficas sugestivas, ou então, haja evidência angiográfica ou necropsica de oclusão coronária.
Atendendo à cinética quer da troponina I quer da troponina T plasmáticas, que podem permanecer em circulação durante 5 e 10 dias, respectivamente, o diagnóstico de re-enfarte pode ser um desafio. Na mais recente redefinição de EAM, os autores (75) sugerem o uso duma percentagem de 20% no aumento da troponina relativamente ao valor prévio para se considerar re-enfarte.
Outro desafio diagnóstico potencial é a identificação de um síndrome coronário agudo na presença de outras doenças (cardíacas ou não) que possam contribuir também para a elevação da troponina I plasmática, à luz do que se discutiu no ponto 6.1. Na situação particular da insuficiência renal crónica, os doentes apresentam frequentemente (em percentagens á volta dos 53%) níveis persistentemente elevados (mas estáveis) de troponina plasmática, tendo sido sugerido o uso de um valor cutoff mais alto nestes doentes, para diagnóstico de enfarte agudo do miocárdio (76). Até porque, mesmo nestas circunstâncias, a troponina I mostrou ter maior sensibilidade e especificidade que a CK-MB (77).
Por tudo isto, importa lembrar que a troponina cardíaca (I ou T) é um marcador necessário para o diagnóstico de enfarte do miocárdio, mas não suficiente.
Como nota final, importa esclarecer que, no caso particular da troponina I, nenhum dos testes actualmente presentes no mercado, cumpre os critérios do consenso (75, 78, 79) da ACC/ESC (em termos de precisão) para diagnóstico de enfarte agudo do miocárdio, uma vez que apresentam coeficientes de variação superiores a 10% para valores equivalentes ao percentil 99 duma população de referência. Todavia, com o Architect STAT da Abbott, a TpI mostrou ter sensibilidade, especificidade e valor preditivo negativo elevados, no contexto de enfarte agudo do miocárdio, sobressaindo relativamente aos seus congéneres.
6.3 - Uso da troponina I como factor de prognóstico
6.3.1 - No contexto de síndromes coronários agudos e doença cardíaca isquémica estável
Tal como referido acima relativamente ao diagnóstico de enfarte agudo do miocárdio, o prognóstico de doentes com síndromes coronários agudos depende sobretudo da troponina, preterindo os antigos marcadores bioquímicos de necrose miocárdica. De facto, Newby e colegas (80) mostraram que, em doentes com EAM sem supradesnivelamento do segmento ST, um valor elevado de troponina cardíaca se associava a um elevado risco de morte intra-hospitalar, independentemente do valor da Ck-MB.
De uma forma geral, a literatura é razoavelmente condizente quanto ao valor prognóstico da troponina I em doentes com síndromes coronários agudos, quer em termos de risco de eventos cardiovasculares quer em termos de mortalidade. Dokainish et al. (81) num subestudo do TIMI-18 demonstraram que as elevações da TpI em doentes com síndromes coronários agudos se associavam a maior risco de morte e re-enfarte, mesmo naqueles doentes sem doença coronária significativa. E num estudo recente (82), Eggers et al. mostraram que uma troponina I persistentemente elevada (> 0,01 ng/ml) após síndromes coronários agudos identificava um grupo de doentes com elevado risco cardiovascular, caracterizados por uma doença coronária mais grave e complexa, bem como por maior disfunção ventricular esquerda.
No caso particular do enfarte agudo com supra-desnivelamento do segmento ST alguns autores (83) mostraram que níveis elevados de troponina I à admissão se correlacionavam de forma significativa e independente com o risco de falência da trombólise e mortalidade aos 30 dias, podendo reflectir a presença de um trombo mais resistente à fibrinólise ou à angioplastia coronária primária, não se podendo excluir uma maior disfunção ou lesão microvascular, que pudessem contribuir secundariamente para uma falência da trombólise e aumento da mortalidade a curto prazo. Outros autores, porém, verificaram que os biomarcadores bioquímicos à admissão tinham poucoimpacto na predição de eventos cardiovasculares a longo prazo (84), e que a troponina I poderia ter interesse prognóstico, apenas se medida às 42h após o evento (85), sendo nesse caso um marcador preciso da extensão do enfarte (à semelhança da Ck-MB máxima) e um indicador da presença de obstrução microvascular.
Relativamente aos restantes síndromes coronários agudos, os estudos são um pouco mais conformes, embora versem mormente no impacto prognóstico a curto prazo. Há mais de uma década atrás Antman et al. (86) constataram que o aumento da troponina I no contexto de enfarte do miocárdio (então designado por enfarte sem ondas Q) ou angina instável identificava um grupo de doentes com elevado risco cardiovascular, predizendo a mortalidade aos 42 dias após o evento. Posteriormente, Agewall et al. (87) demonstraram que níveis detectáveis de TpI (ainda que abaixo do valor considerado cutoff para diagnóstico de enfarte agudo do miocárdio), em doentes admitidos numa unidade coronária, prediziam significativamente a mortalidade aos 40 dias. Finalmente, no estudo de Apple et al. (22) referido anteriormente neste texto, foi demonstrado que pequenas subidas de TpIc tinham implicações prognósticas, tendo-se documentado um maior risco de eventos adversos no grupo de doentes que tinham TpIc detectável (ainda que abaixo do percentil 99), em comparação com aqueles com valores indetectáveis.
Quando da revascularização coronária, quer cirúrgica quer percutânea, ocorre sempre libertação de pequenas quantidades de troponina cardíaca, mesmo que o procedimento decorra conforme protocolado e na ausência de enfarte do miocárdio tipo IV ou tipo V. Esta troponinemia relacionada com a intervenção coronária pode ser usada peri-procedimento para elucidar acerca da agressão miocárdica concomitante. De facto, Onorati (88) mostrou que a TpI in tra-operatória podia permitir a quantificação do grau de lesão miocárdica, ainda melhor que o lactado, usado até então como marcador metabólico da perfusão coronária. Para obviar o problema da pequena amplitude na elevação da troponina I, o grupo de Burnetti (89) criou um novo marcador – a razão entre a troponina I máxima e a troponina I basal – tendo-se verificado que valores elevados desta razão (> 1) prediziam significativamente a ocorrência de eventos cardiovasculares adversos major, 6 meses após o procedimento, mesmo após ajuste para idade, sexo, factores de risco cardiovasculares, tipo de síndrome coronário e troponina I máxima.
Não obstante, e à semelhança do que passa com os síndromes coronários agudos, as pequenas elevações da troponina I relacionadas com os procedimentos de revascularização coronária, não parecem ter qualquer impacto no prognóstico a longo prazo (1 ano após), em termos de mortalidade ou ocorrência de eventos cardiovasculares major (90).
Recentemente, Omland et al. estudaram 3593 doentes com doença coronária estável, utilizando um um novo método de medição da Troponina T, altamente sensível (91). As concentrações crescentes de troponina T associaram-se à incidência de mortalidade cardiovascular e à incidência de insuficiência cardíaca mas não à incidência de enfarte do miocárdio (91).
6.3.2 - No contexto de outras situações patológicas
A par do que acontece nos doentes com síndromes coronários agudos ou com doença cardíaca isquémica estável, pequenas subidas da TpI também acarretam significado prognóstico noutros estados patológicos e mesmo em indivíduos ditos saudáveis. Far-se-á de seguida uma breve revisão do conhecimento actual relativo a este assunto.
6.3.2.1 - Em doentes críticos
A presença de níveis detectáveis de troponina cardíaca (entre 0,4 – 3,6 µg/L) em doentes críticos (sem síndromes coronários agudos) admitidos em Unidades de cuidados intensivos (UCI) é relativamente frequente (variando entre 15,3% e 25%, conforme os estudos (92)), sobremaneira no contexto de sépsis/SRIS (síndrome de resposta inflamatória sistémica), trauma, insuficiência respiratória aguda e choque, tendo sido demonstrado, por diversos autores (51,93,94), que constitui um factor de mau prognóstico, independentemente da causa.
King et al. (95) verificaram, numa pequena coorte (n = 128) prospectiva de doentes consecutivamente admitidos numa UCI médica que aqueles que tinham troponina I positiva (> 0,7 ng/ml) apresentavam maior mortalidade (odds ratio (OR) = 7,0, p < 0,001) aos 28 dias, maior pontuação no APACHE (Acute Physiology and Chronic Health Evaluation) II e uma maior taxa de falência multiorgânica e sépsis. Todavia, quando ajustado para oAPACHE II, a positividade da TpI não acrescentava valor preditivo para a mortalidade, relativamente àquele fornecido pelo APACHE II.
Mais recentemente, no estudo de Stein et al. (96) demonstrou-se que valoresintermédios (0,1–1,49 ng/ml) de TpI (ADVIA Centaur da Bayer, com limiar de detecção de 0,09 ng/ml), constituíam factor preditivo independente (tal como o SAPS-II) para a mortalidade intra-hospitalar de doentes críticos não cirúrgicos.Aexistência de doença coronária prévia não alterou significativamente o valor da TpI, e os autores especularam que esta subida da TpI reflectia provavelmente a maior gravidade da condição clínica subjacente. De facto os doentes com valores intermédios de TpI eram mais velhos e pontuavam mais no SAPS-II quando comparados com aqueles que tinham TpI < 0,1 ng/ml.
O pior prognóstico dos doentes críticos sem SCA que apresentem troponinemia detectável pode deverse também a uma maior resposta inflamatória sistémica e / ou a complicações cardiovasculares. De facto, já Ammann et al. (97), num pequeno estudo prospectivo, tinham verificado que doentes com valores detectáveis de troponina tinham maior probabilidade de morrer aos 30 dias, mostrando menor fracção de ejecção do ventrículo esquerdo (feVE) e níveis mais elevados de marcadores inflamatórios (TNF-α, IL-6).
Srivathsan et al. (98) estudaram oimpacto cardiovascular de pequenas elevações da TpI (na presença de CK-MB normal) em indivíduos gravemente doentes internados, e verificaram que aqueles que possuíam TpI detectável apresentavam maior taxa de mortalidade e maior risco de enfarte do miocárdio ou re-admissão hospitalar por motivos cardiovasculares, independentemente do motivo de internamento inicial.
6.3.2.2 - Sépsis
A sépsis e o choque séptico são as principais causas de morbi-mortalidade dos doentes críticos internados em UCIs médicas, presumivelmente pelas complicações cardiovasculares inerentes, tal como já explorado previamente neste texto.
Alguns autores (95) mostraram que doentes com sépsis que mostravam valores positivos de troponina cardíaca tinham tendência a ter uma fracção de ejecção ventricular esquerda menor, que aqueles com troponina negativa, podendo este ser um dos factores pelos quais os doentes sépticos com troponina positiva têm apresentado pior prognóstico que aqueles com troponina negativa.
Em conformidade com o exposto, Mehta et al. (99) constataram que níveis elevados de TpI em doentes com sépsis ou choque séptico constituíam um marcador independente de mau prognóstico, predizendo maior gravidade da sépsis bem como maior mortalidade (56% vs. 24%, p = 0,04), comparativamente com os doentes negativos para TpI.
6.3.2.3 - Endocardite infecciosa
Na literatura encontram-se várias descrições relativas ao aumento da troponina I plasmática no contexto da endocardite infecciosa, mas ainda se sabe pouco quanto às implicações prognósticas da detecção desse biomarcador neste tipo particular de doentes.
Recentemente, Purcell et al. (100), estudando uma coorte de 83 doentes com endocarditeinfecciosa, demonstraram que aqueles que possuíam níveis elevados de TpI, tinham maior tendência a desenvolver disfunção sistólica do ventrículo esquerdo e insuficiência renal, bem como uma maior prevalência de complicações por formação de abcessos. Nesta sequência, especulou-se que o aumento da TpI em doentes com endocardite infecciosa podia servir como um marcador de infecção mais grave, envolvendo o próprio miocárdio.
6.3.2.4.- Tromboembolismo pulmonar (TEP) agudo
Tal como analisado previamente neste texto, é frequente encontrar pequenas elevações da troponina I plasmática em doentes com TEP agudo, tendo sido já descrito um aumento da mortalidade intra-hospitalar em doentes com embolia pulmonar e níveis elevados de troponina I (101).
Recentemente, Kline et al. (62) comparando 8 potenciais marcadores bioquímicos de disfunção ventricular direita no âmbito do TEP agudo, demonstraram que, em doentes com tromboembolismo pulmonar sub-massivo, apenas a troponina I e o BNP constituem factores de prognóstico significativos, ainda que pouco robustos (área abaixo da curva ROC de 0,71). Usando os cutoffs de 100 pg/ml e 0,1 ng/ml, respectivamente para o BNP e TpI, aqueles autores verificaram que níveis superiores a estes valores prediziam significativamente a hipocinésia ventricular direita, com discreta supremacia do BNP por também predizer a mortalidade.
6.3.2.5. - Insuficiência cardíaca congestiva
Horwich et al. (102) verificaram que níveis elevados de troponina I cardíaca em doentes com IC crónica constituíam um marcador de mau prognóstico. E, mais recentemente, o grupo de Fonarow (103), numa subanálise do estudo ADHERE, avaliando a performance de vários marcadores bioquímicos, verificou que níveis elevados de troponina I (≥ 1,0 ng/ml) e BNP (≥ 840 pg/ ml), em doentes hospitalizados por IC agudizada, eram factores preditivos independentes de mortalidade intrahospitalar (OR = 2,09 e 2,41, respectivamente, com p < 0,0001). Importa ainda acrescentar, que o uso de vários marcadores de risco cardiovascular em simultâneo aumenta significativamente a acuidade na estratificação do risco cardiovascular de doentes com IC crónica. De facto, numa coorte de 152 doentes com IC grave, Yin et al. (104) mostraram que aqueles doentes com três marcadores [troponina I, BNP e proteína C reactiva de alta sensibilidade (PCRas)]positivos tinham 23,4 vezes maior risco de sofrer eventos cardiovasculares que aqueles que os tinham negativos, enquanto aqueles que mostrassem apenas um marcador positivo tinham um risco apenas 2,7 vezes superior aos negativos.
À semelhança do que se passa com a IC crónica, também na IC aguda foi demonstrado o valor prognóstico da troponina cardíaca (105). You e colaboradores (106), no âmbito do estudo EFFECT, investigando a associação entre a troponina I e a mortalidade global (independentemente da causa), verificaram que a troponina I se correlacionava linearmente com a mortalidade, e que um valor superior a 0,5 µg/L era factor preditivo independente de mortalidade (Hazard Ratio (HR) = 1,49 com Intervalo de Confiança a 95% (IC95%) 1,25 – 1,77).
6.3.2.6 - Hemorragia subaracnoideia
A hemorragia subaracnoideia (HSA) aneurismática associa-se a uma série de complicações cardíacas (particularmente alterações da motilidade da parede), ainda que transitórias, pensando-se que o principal mecanismo seja uma descarga catecolaminérgica quando da ruptura do aneurisma, com subsequente lesão miocárdica.
Naidech et al. (107) investigaram a troponina I como método de rastreio de complicações cardiopulmonares em doentes com HSA. Para isso, estudaram uma coorte de 441 doentes com HSA, procedendo a doseamentos seriados da troponina I naqueles que apresentassem alterações electrocardiográficas (prolongamento do QT, ondas Q, alterações do segmento ST ou inversão das ondas T) ou clínica sugestiva de disfunção cardíaca (hipotensão ou hipertensão arterial, edema agudo do pulmão, disritmias cardíacas ou dor torácica). Verificaram que os valores máximos de TpI eram factores preditivos de risco elevado de hipotensão severa (com necessidade de tratamento vasopressor), edema pulmonar, disfunção sistólica do ventrículo esquerdo (VE) e isquemia cerebral prolongada por vasospasmo. Além disso, a TpI máxima também mostrou uma associação significativa com risco elevado de morte ou incapacidade severa à data da alta. Todavia, este efeito perdia significância aos 3 meses após alta hospitalar.
Tanabe et al. (108) estudaram prospectivamente 103 doentes com HSA tendo constatado que aqueles que apresentavam valores elevados de troponina I (≥ 1,0 ng/ml) exibiam lesões neurológicas mais graves, tinham tempos de internamento em UCI mais longos, e mostravam maior disfunção cardíaca sistólica e diastólica, bem como um maior grau de congestão pulmonar. E mesmo naqueles doentes com pequenas elevações de TpI ( 0,1 – 1,0 ng/ml) verificaram um aumento significativo de disfunção ventricular diastólica e congestão pulmonar. Não obstante, estas alterações eram transitórias resolvendo em 5 a 10 dias após a HSA. De forma similar, o grupo de Sandhu (109), usando uma coorte mais heterogénea (acidente vascular cerebral isquémico, hemorragia intracerebral e hemorragia subaracnoideia) mostrou que os doentes com HSA e níveis elevados de TpI (≥ 0,4ng/ml) tinham uma mortalidade intra-hospitalar 3,6 vezes maior que aqueles com TpI normal (< 0,4 ng/ml).
A troponina cardíaca parece, portanto, ser um marcador significativo do prognóstico intra-hospitalar de doentes com HSA, faltando estudar qual o seu impacto em termos de prognóstico a longo prazo, bem como a exequibilidade da implementação de uma estratégia de estratificação de risco cardiovascular usando a TpI.
6.3.2.7 - Estenose aórtica
Em doentes com estenose aórtica (EA), a existência de disfunção sistólica do ventrículo esquerdo (VE) e insuficiência cardíaca prediz um mau prognóstico, incluindo o eventual insucesso após cirurgia de substituição valvular (110).
Nunes et al. (58) estudaram 25 doentes com doença valvular aórtica estável, testemunhando neste grupo, valores médios de troponina I significativamente superiores àqueles dos indivíduos controlo, e verificaram que pequenos aumentos na troponina I plasmática se associavam significativamente a uma parede ventricular esquerda mais espessada e uma pressão arterial pulmonar mais elevada. Dois anos depois, Kupari et al. (111) tentaram analisar se o doseamento seriado de troponina I plasmática em doentes com estenose aórtica permitia identificar a destruição silenciosa dos miocardiócitos que antecede a IC associada à EA, e notaram que grande parte dos doentes com EA tinham níveis detectáveis de TpI, e que pelo menos 1/5 tinha níveis elevados (> 14 ng/L), correlacionando-se significativamente com disfunção sistólica do VE. Face a estes resultados os autores especularam a utilidade de medições seriadas da TpI plasmática como marcador de disfunção ventricular, permitindo a implementação de medidas terapêuticas mais agressivas com o objectivo de prevenir o desenvolvimento de IC franca.
6.3.2.8 -No pós-operatório de cirurgias cardíaca e não cardíaca
É frequente encontrar subidas da troponina plasmática após qualquer cirurgia cardíaca, reflectindo provavelmente lesão cardíaca peri-operatória. Adabag et al. (112) estudaram o significado prognóstico da troponina I pós-operatória em doentes submetidos a cirurgia de revascularização coronária (CRC) (n = 696) ou substituição valvular (CSV) (n = 490), tendo encontrado um aumento de 40% e 30% na probabilidade de morrer durante a CRC e CSV, respectivamente, por cada aumento de 50 ng/ml da troponina I plasmática. Alguns autores (113), estudaram especificamente uma coorte de mulheres pós-menopausicas, chegando às mesmas conclusões: níveis elevados de troponina I, no 1º dia pós-operatório, são preditivos de eventos cardiovasculares major. De forma similar, Nesher et al. (114) investigaram a correlação entre a troponina T pós cirurgia cardíaca e a ocorrência de eventos cardiovasculares major (ECM), concluindo que valores de troponina T superiores a 0,8 µg/L se correlacionavam significativamente com uma maior frequência de ECM nos 30 dias seguintes à cirurgia cardíaca, em doentes sem história pré-operatória de enfarte do miocárdio.
Relativamente à cirurgia vascular major (ex. correcção de aneurisma da aorta abdominal), Bolliger et al. (115) mostraram que o doseamento da troponina I pós-cirurgia em conjunto com a medição do BNP pré-cirurgia, fornecia informação prognóstica adicional e significativa em termos de eventos cardiovasculares major e mortalidade. Por sua vez, Mantha et al. (116) comprovaram a relação custo-eficácia da implementação de uma estratégia de vigilância intensiva pós-operatória (com determinações diárias de TpI nos primeiros 4 dias) em doentes submetidos a cirurgia electiva da aorta abdominal.
6.3.2.9 - Insuficiência renal crónica
Os doentes cominsuficiência renalcrónica apresentam frequentemente níveis detectáveis de troponina, tendo já sido demonstrado que estes constituem um factor preditivo robusto da mortalidade a médio – prazo (117) naqueles doentes cominsuficiência renalcrónica estável, sobretudo se associado à proteína C reactiva (118). Não obstante, o valor prognóstico da troponina deve ser sempre considerado no contexto da gravidade da insuficiência renal. De facto, Melloni e colaboradores (119) mostraram que em doentes com SCA sem supra-desnivelamento do segmento ST, e função renal normal ou com insuficiência renal ligeira, a mortalidade era baixa independentemente do valor da troponina, mas naqueles com insuficiência renal moderada, a mortalidade já aumentava gradualmente conforme o acréscimo da troponina I ou T, e no caso dos doentes com insuficiência renal grave, apenas a troponina T adicionava valor preditivo da mortalidade relativamente à severidade da falência renal.
6.3.2.10 -No contexto de quimioterapia com antraciclinas
De todos os citostáticos conhecidos as antraciclinas constituem o grupo de fármacos com maior risco de cardiotoxicidade (120), tendo-se vindo a investigar qual o melhor método de monitorização cardiovascular a implementar nos doentes oncológicos sob este tipo de terapêutica.
Horacek et al. (121) pesquisaram a utilidade da troponina cardíaca na vigilância da toxicidade cardiovascular das antraciclinas, e observaram que apenas a troponina I era detectável no dia seguinte à primeira e à última sessão de quimioterapia, mostrando, portanto, superioridade relativamente à troponina T na detecção precoce da lesão cardíaca induzida por aquela classe de medicamentos. Os doentes que apresentavam troponina I positiva durante o tratamento com antraciclinas, sofriam, à posteriori, um decréscimo significativamente maior na fracção de ejecção do ventrículo esquerdo, quando comparados com os doentes negativos para esse biomarcador. Perante estes resultados, os autores admitiram que a troponina I poderia vir a ser usada na identificação daqueles doentes com maior risco de desenvolver futuramente cardiomiopatia induzida pelas antraciclinas, permitindo intervenção precoce.
Pelo que aqui se descreveu fica claro que a troponina I além de ser um importante marcador bioquímico usado no diagnóstico do enfarte agudo do miocárdio, constitui, de forma similar, um factor de prognóstico bastante significativo, quer em doentes com síndromes coronários agudos, quer naqueles com doenças cardíacas não isquémicas ou outros estados patológicos, predizendo mormente a mortalidade e eventos cardiovasculares major.
Acresce, ainda, um potencial papel imunomodelador da troponina I, podendo contribuir para a patofisiologia inflamatória das doenças cardiovasculares, com desestabilização quer do doente quer da placa aterosclerótica.
Efectivamente, no estudo experimental de Göser et al. (122), verificou-se que a troponina I (mas não a T) induzia uma resposta auto-imune (com produção de anticorpos anti-TpI) e agravava a extensão de um subsequente enfarte do miocárdio, bem como a reacção inflamatória e a disfunção ventricular dele procedentes.
6.3.2.11 - Doença pulmonar obstrutiva crónica
A literatura é unânime em revelar que os doentes com doença pulmonar obstrutiva crónica (DPOC) apresentam frequentemente níveis detectáveis de troponina cardíaca. Contudo, escasseiam estudos quanto ao significado prognóstico desses aumentos da troponina no contexto das agudizações da DPOC, tendo-se encontrado apenas 3 trabalhos, conforme exposto de seguida.
Baillard et al. (123) investigaram a incidência e o valor prognóstico de elevações da troponina I (TpI) em 71 doentes admitidos em unidades de cuidados intensivos (UCI) por agudização severa da DPOC, tendo constatado positividade para TpI (definida como > 0,5ng/ml) em 18%. Os doentes com níveis de TpI superiores a 0,5 ng/ml, pontuavam mais no SAPS II, tinham PaO2 mais alta à admissão e apresentavam uma probabilidade de mortalidade intra-hospitalar significativamente maior (p = 0,002) que os que não tinham troponina detectável.
Harvey et al. (124) estudaram retrospectivamente 235 doentes admitidos no hospital por agudização da DPOC que tivessem determinação da troponina cardíaca à admissão, tendo verificado que cerca de 25% apresentavam níveis elevados de troponina cardíaca, sendo tendencialmente mais velhos, com SatO2 mais baixas, valores mais baixos de pH e PaCO2 mais elevada à admissão, bem como um tempo de internamento mais prolongado. Em face dos resultados, os autores postularam que a detecção da troponina cardíaca em doentes com DPOC agudizada pode reflectir uma maior gravidade da agudização.
Mais recentemente, Brekke et al. (125), estudando retrospectivamente uma coorte de 396 doentes admitidos no hospital por agudização da DPOC, verificaram um discreto mas significativo aumento da mortalidade extrahospitalar naqueles com níveis elevados de Troponina T (> 0,04 ng/ml).
Martins et al. estudaram 173 doentes com agudizações de DPOC e com medição de troponina I (126). Cerca de 70% dos casos tinham um valor de troponina I superior ao percentil 99. A troponina I correlacionou-se com a necessidade de ventilação não-invasiva, bem como com a sobrevida aos 18 meses.
7 - CONCLUSÕES
As troponinas cardíacas, e em especial a troponina I, são proteínas que funcionam como marcadores de lesão miocárdica, sendo altamente específicas e selectivas, e apesar do seu interesse inicial se ter prendido com o diagnóstico do enfarte agudo do miocárdio, actualmente esse seu papel não é tão linear, tendo havido mesmo quem sugerisse revisão dos conceitos de enfarte e pseudo-enfarte do miocárdio. Efectivamente, são muitas as situações em que se pode detectar troponina no plasma, sendo importante que se mantenha um juízo crítico em cada circunstância.
REFERÊNCIAS
1 -Guyton AC, Hall JE. Heart muscle; the heart as a pump. In: Textbook of Medical Physiology, 9th ed, WB Saunders Company 1996;pp.107-10.
2 -Kobayashi T, Jin L, de Tombe PP. Cardiac thin filament regulation. Pflugers Arch 2008;457:37-46.
3 -Tobacman LS, Butters CA. A new model of cooperative myosine-thin filament binding. J Biol Chem 2000;275: 27587-93.
4 -Lechman W, Hatch V, Korman V, et al. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J Mol Biol 2000;302:593.
5 -Rakoczy AG, Engel P, Xu C, et al. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J Mol Biol 2008;379:929-35.
6 -Tregar RT, Reedy MC, Goldman YE, Taylor KA, Winkler H, Franzini AC, et al. Cross-brigde number, position and angle in target zones of cryofixed isometrically active inserct flight muscle. Biophys J 2004;86:3009-19.
7 -Maytum R, Westerdorf B, Jaquet K, Geeves MA. Differential regulation of the actomyosin interaction by skeletal and cardiac troponin isoforms. J Biol Chem 2008;278: 6696-701.
8 -Hoffman RMB, Sykes BD. Isoform specific variation in the intrinsic disorder of troponin I. Proteins 2008;73:338-50.
9 -Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem Biophys Res Commun 2008;369:8-7.
10 -Sadayappan S, Finley N, Howarth JW, et al. Role of the acidic N’region of cardiac troponin Iin regulating myocardial function. FASEB J 2008;22:1246-57.
11 -Mathur MC, Kobayashi T, Chalovich JM. Negative charges at protein kinase C sites of troponin I stabilize the inactive state of actin. Biophys J 2008;94:542-9.
12 -Genaro A, Correa R, Murphy AM. Is phospholambam or troponin I the “prima-donna” in β-adrenergic induced lusitropy? Circul Res 2007;101:377-86.
13 -Le Moal E, Giuliani I, Bertinchant JP, Polge A, Larue C, Villard-Saussine S. Earlier detection of myocardial infarction by an improved cardiac TpI assay. Clin Biochem 2007; 40:1065-73.
14 -Collinson PO, Boa FG, Gaze DC. Measurement of cardiac troponins. Ann Clin Biochem 2001;38:423-49.
15 -Panteghini M, Bunk DM, Christenson RH, et al. Standardization of troponin I measurement: an update. Clin Chem Lab Med 2008;46:1501-6.
16 -Higgins JP, Higgins JA. Elevation of cardiac troponin I indicates more than myocardial ischemia. Clin Invest Med 2003;26:133-47.
17 -Apple FS, Murakami MM. Serum and plasma cardiac troponin I 99th percentile reference values for 3 2nd generation assays. Clin Chem 2007;53:1558-60.
18 -Venge P, James S, Jansson L, Lindahl B. Clinical performance of two highly sensitive cardiac troponin I assays. Clin Chem 2009;55:109-16.
19 -Hjortshoj S, Dethlefsen C, kristensen SR, Ravkilde J. Improved assay of cardiac troponin I is more sensitive than other assays of necrosis markers. Scan J Clin Lab Invest 2008;68:130-3.
20 -Kavsak PA, MacRae AR, Newman AM, et al. Effects of contemporary troponin assay sensitivity on the utility of the early markers myoglobin and CK-MB isoforms in evaluating patients with possible acute myocardial infarction. Clin Chim Acta 2007;380:213-6.
21 -Wiesner DC, Parsons R, Olson R, Waterston J, Smith C. Enhanced Sensitivity of the ARCHITECT STAT Troponin-I assay at three clinical reference points. American Association for Clinical Chemistry Annual Meeting, Chicago, Illinois, July 23 - 27, 2006.
22 -Apple FS, Smith W, Pearce LA, Ler R, Murakami MM. Use of the Centaur TnI-Ultra assay for detection of myocardial infarction and adverse events in patients presenting with symptoms suggestive of acute coronary syndrome. Clin Chem 2008;54:723-8.
23 -Casals G, Filella X, Bedini JL. Evaluation of a new ultrasensitive assay for cardiac troponin I. Clin Biochem 2007;40:1406-13.
24 -Prontera C, Fortunato A, Storti S, et al. Evaluation of analytical performance of the Siemens ADVIA TnI-Ultra Immunoassay. Clin Chem 2007;53:1722-3.
25 -Zhu Y, Jenkins MM, Brass DA, et al. Heterophilic antibody interference in an ultra-sensitive 3-site sandwich troponin I immunoassay. Clin Chem Acta 2008;395:181-2.
26 -John Todd, Bob Freese, Ann Lu, et al. Ultrasensitive flow-based immunoassay using single-molecule counting. Clin Chem 2007;53:1990-5.
27 -Wu AHB, Agee SJ, Lu QA, Todd J, Jaffe AS. Specificity of a high-sensitivity cardiac troponina I assay using single-molecule-counting technology. Clin Chem 2009;55:196-8.
28 -Reichlin T, Hochholzer W, Basseti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin as says. N Engl J Med 2009;361:858-67.
29 -Pentilla IM, Laatikainen A, Pentilla K, et al. Imprecision of cardiac marker among laboratories on the basis of external quality assurance results: Finnish experience. Scand J Clin Lab Invest 2008;67:507-18.
30 -Katrukha A, Bereznikova A, Pettersson K. New approach to standardization of human cardiac troponin I (cTnI). Scand J Clin Lab Invest 1999;59(suppl 230):124-7.
31 -Cobbaert CM, Weykamp CW, Michielsen ECHJ, Baadenhuijsen H, Van Dieijen-Visser MP. Time-dependent instability of cardiac troponins in human plasma spiked with NIST reference material 2921. Clin Chem 2008;54:2078-89.
32 -Collinson P, Pulkki K, Suvisaari J, et al. How well do laboratories follow guidelines on cardiac markers? The cardiac marker guideline uptake in Europe study. Clin Chem 2008; 54:448-52.
33 -Apple FS, Parvin CA, Buechler KF, et al. Validation of the 99th percentile cutoffindependent of assayimprecision (CV) for cardiac troponin monitoring for ruling out myocardial infarction. Clin Chem 2005;51:2198-200.
34 -Tze-Kiong ER, Gines MAR, Yuh-Jyh Jong, et al. Falsely elevated cardiac troponin I attributed to inappropriate dilution material. Am J Emerg Med 2009;27:123-4.
35 -Hejl CG, Astier HT, Ramirez JM. Prevention of preanalytical false-positive increases of cardiac troponin I on the Unicel Dxl 800 analyzer. Clin Chem Lab Med 2008; 48:1789-90.
36 -Daves M, Trevisan D, Cemin R. Different collection tubes in cardiac biomarkers detection. J Clin Lab Anal 2008;22:391-4.
37 -Prince AM, Brotman B, Jass D, Ikram H. Specificity of the direct solid-phase radioimmunoassay for detection of hepatitis-B antigen. Lancet 1973;1:1346-50.
38 -Yeo KT, Storm CA, Li Y, et al. Performance of the enhanced Abbott AxSym cardiac troponin I reagent in patients with heterophilic antibodies. Clin Chem Acta 2000;292:13-23.
39 -Lippi G, Tessitore N, Montagnana M, Salvagno GL, Lupo A, Guidi GC. Influence of sampling time and ultrafiltration coefficient of the dialysis membrane on cardiac troponin I and T. Arch Pathol Lab Med 2008;132:72-6.
40 -Melanson SEF, Conrad MJ, Mosammaparast N, Jarolim P. Implementation of a highly sensitive cardiac troponin I assay: test volumes, positivity rates and interpretation of results. Clin Chim Acta 2008;395:57-61.
41 -Hochholzer W, Buettner HJ, Trenk D, et al. New definition of myocardial infarction: impact on long-term mortality. Am J Med 2008;121:399-405.
42 -Wu AHB, Lu QA, Todd J, Moecks J, Wians F. Short and long-term biological variationin cardiac troponin I measured with a high-sensitivity assay: implications for clinical practice: Clin Chem 2009;55:52-8.
43 -Kurihara T, Yanagida A, Yokoi H, et al. Evaluation of cardiac assays on a benchtop chemiluminescent enzyme immunoassay analyzer, PATHFAST. Anal Biochem 2008;375:144-6.
44 -Cho IH, Paek EH, Kim YK, Kim JH, Paek SH. Chemiluminometric enzyme-linked immunosorbent assays (ELISA)-on-a-chip biosensor based on cross-flow chromatography. Anal Chim 2009;632:247-55.
45 -Singh J, Akbar MS, Adabag S. Discordance of cardiac troponin I assays on the point-of-care i-STAT and Architect assays from Abbott diagnostics. Clin Chim Acta 2009;403:259-60.
46 -Remppis A, Scheffold T, Greten J, et al. Intracellular compartmentation of troponin T – release kinetics after global – ischemia and calcium paradox in the isolated – perfused rat-heart. J Mol Cell Cardiol 1995;27:793-803.
47 -Khan IA, Tun A, Wattanasauwan N, et al. Elevation of serum cardiac troponin I in noncardiac and cardiac diseases other than acute coronary syndromes. Am J Emerg Med 1999;17:225-9.
48 -Pham MX, Whooley MA, Evans Jr GT, et al. Prognostic value of low-level cardiac troponin-I elevations in patients without definite acute coronary syndromes. Am Heart J 2004;148:776-82.
49 -Mahajan N, Mehta Y, Rose M, ShaniJ, Lichstein E. Elevated troponinlevelis not synonymous with myocardialinfarction. Int J Cardiol 2006;11:442-9.
50 -Jeremias A, Gibson CM. Narrative review: alternative causes for elevated cardiac troponin levels when acute coronary syndromes are excluded. Ann Intern Med 2005;142:786-91.
51 -Lazzeri C, Bonizzoli M, Cianchi G, Gensini GF, Peris A. Troponin I in the intensive care unit setting: from the heart to the heart. Intern Emerg Med 2008;3:9-16.
52 -Blich M, Sebbag A, Attias J, Aronson D, Markiewickz W. Cardiac troponin I elevation in hospitalized patients without acute coronary syndromes. Am J Cardiol 2008;101: 1384-8.
53 -Zellweger MJ, Schaer BA, Cron TA, Pfisterer ME, Osswald S. Elevated troponin levels in the absence of coronary artery disease after supraventricular tachycardia. Swiss Med WKLI 2003;133:439-41.
54 -Bakshi TK, Choo MKF, Edwards CC, et al. Causes of elevated troponin I with a normal coronary angiogram. Int Med J 2002;32:520-5.
55 -Logeart D, Beyne P, Cusson C, et al. Evidence of cardiac myolysis in severe non ischemic heart failure and the potential role of increased wall strain. Am Heart J 2001; 141:247-53.
56 -Nunes JPL. Cardiac troponin I in systemic diseases. A possible role for myocardial strain. Rev Port Cardiol 2001; 20:785-8.
[ Links ]57 -Logeart D, Beyne P, Cusson C, et al. Evidence of cardiac myolisis in severe nonischemic heart failure and potential role of increased wall strain. Am Heart J 2001;141:247-53.
58 -Angheloiu GO, Dickerson RP, Ravakhah K. Etiology of troponin I elevation in patients with congestive heart failure and low clinical suspicion of myocardial infarction. Ressuscitation 2004;63:195-201.
59 -Nunes JP, Mota Garcia JM, Farinha RM, et al. Cardiac troponin I in aortic valve disease. Int J Cardiol 2003;89: 281-5.
60 -Feng J, Schaus BJ, Fallavolita JA, Lee TC, Canty Jr JM. Preload induces troponin I degradation independently of myocardial ischemia. Circulation 2001;103:2035-7.
61 -Hessel MHM, Atsma DE, van der Valk EJM, Bax WH, Schalij MJ, van der Laarse A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Eur J Physiol 2008;455:979-86.
62 -Kline JA, Zeitouni R, Marchick MR, Hernandez-Nino J, Rose GA. Comparison of 8 biomarkers for prediction of right ventricular hypokinesis 6 months after submassive pulmonary embolism. Am Heart J 2008;156:308-14.
63 -Nunes JPL, Magalhães D, Dias P, Faria DB. Troponin I elevation after pericardiocentesis for cardiac tamponade: a role for myocardial strain? Int J Cardiol 2001;81:277-8.
64 -Pateron D, Beyne P, Laperche T, et al. Elevated circulating cardiac troponin I in patients with cirrhosis. Hepatology 1999;29:640-43.
65 -Chan AOK, Chan JPS, Choi KL, Shek CC. A patient with an increased troponin level without evidence of ischaemic cardiac injury. Hong Kong Med J 2004;10:277-9.
66 -Parmar MS. We need to address the issue of “mild troponinaemia”. BMJ 2009;338:b 769.
67 -Lee SH, Yoon SB, Jung JH, et al. Prognostic factors in patients with minor troponin-I elevation but without acute myocardial infarction. Coron Artey Dis 2006;17:249-53.
68 -Collinson PO, Gaze DC. Biomarkers of Cardiovascular Damage. Med Princ Pract 2007;16:247-61.
69 -Collinson PO, Gaze DC. Biomarkers of Cardiovascular Damage and Dysfunction – An Overview. Heart, Lung and Circulation 2007;16:S7-S82.
70 -Apple FS, Smith SW, Pearce LA, Murakami MM. Assessment of the Multiple-Biomarker Approach for Diagnosis of MyocardialInfarctionin Patients Presenting with Symptoms Suggestive of Acute Coronary Syndrome. Clin Chem 2009; 55:93-100.
71 -Ilva T, Lund J, Porela P, et al. Early markers of myocardial injury: cTnI is enough. Clin Chim Acta 2009;400:82-5.
72 -Topol EJ, Bates ER, Walton JA Jr, et al. Community hospital administration of intravenous tissue plasminogen activator in acute myocardialinfarction:improved timing, thrombolytic efficacy and ventricular function. J Am Coll Cardiol 1987; 10:1173-7.
73 -Stubbs P, Collinson P, Moseley D, Greenwood T, Noble M. Prognostic significance of admission troponin T concentration in patients with myocardial infarction. Circulation 1996;94:1291-7.
74 -Ohman EM, Armstrong PW, Christenson RH, et al. Cardiac troponin T levels for risk stratification in acute myocardial ischemia. N Engl J Med 1996;335:1333-41.
75 -Thygesen K, Alpert JS, White HD. Joint ESC /ACCF/AHA/ WHF task force for the redefinition of myocardial infarction. Universal definition of myocardial infarction. Eur Heart J 2007;28:2525-38; Circulation 2007;116:2634-53; J Am Coll Cardiol 2007;50:2173-95.
76 -Van Lente F, McErlean ES, DeLuca SA, Peacock WF, Rao JS, Nissen SE. Ability of troponins to predict adverse outcomes in patients with renal insufficiency and suspected acute coronary syndromes: a case-matched study. J Am Coll Cardiol 1999;33:471-78.
77 -Martin GS, Becker BN, Schulman G. Cardiac troponin-I accurately predicts myocardial injury in renal failure. Nephrol Dial Transplant 1998;13:1709-12.
78 -Alpert JS, Thygesen K, Jaffe A, White HD. The universal definition of myocardial infarction: a consensus document. Heart 2008;94:1335-41.
79 -HublW, DemantT, Gladrow E. Evaluation of theARCHITECT STAT troponin-I assay. Clin Lab 2005;51:251-5.
80 -Newby LK, Roe MT, Chen AY, et al. Frequency and clinical implications of discordant creatine kinase – MB and troponin measurements in acute coronary syndromes. J Am Coll Cardiol 2006;47:312-8.
81 -Dokainish H, Pillai M, Murphy SA, et al. Prognostic Implications of Elevated Troponin in Patients with Suspected Acute Coronary Syndrome but no Critical Epicardial Coronary Disease. A TACTICS-TIMI-18 Substudy. J Am Coll Cardiol 2005;45:19-24.
82 -Eggers KM, Lagerqvist B, Oldgren J, et al. Pathophysiologic mechanism of persistent cardiac troponin I elevation in stabilized patients after an episode of acute coronary syndrome. Am Heart J 2008;156:588-94.
83 -Foussas SG, Zairis MN, Makrygiannis SS, et al. The significance of circulating levels of both cardiac troponin I and high-sensitivity C reactive protein for the prediction of intravenous thrombolysis outcome in patients with ST-segment elevation myocardial infarction. Heart 2007;93:952-6.
84 -Jeong YH, Lee SW, Lee CW, et al. Biomarkers on admission for the prediction of cardiovascular events after primary stenting in patients with ST-elevation myocardial infarction. Clin Cardiol 2008;31:572-9.
85 -Younger JF, Plein S, Barth J, et al. Troponin I concentration 72h after myocardial infarction correlates with infarct size and presence of microvascular obstruction. Heart 2007; 93:1547-51.
86 -Antman EM, Tanasijevic MJ, Thompson B, et al. Cardiac-specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. N Engl J Med 1996;335:1342-9.
87 -Agewall S, Olsson T, Löwbeer C. Usefulness of troponin levels below the diagnostic cut-off level for acute myocardial infarction in predicting prognosis in unselected patients admitted to the coronary care unit. Am J Cardiol 2007;99:1357-9.
88 -Onorati F, Cristodoro L, Caroleo S, et al. Troponin I and lactate from coronary sinus predict cardiac complications after myocardial revascularization. Ann Thorac Surg 2007; 83:1016-23.
89 -Burnetti ND, Quagliara D, Di Biase M. Troponin ratio and risk stratification in subjects with acute coronary syndrome undergoing percutaneous coronary intervention. Eur J Int Med 2008;19:435-42.
90 -Labriolle AD, Lemesle G, Bonello L, et al. Prognostic significance of small troponin I rise after a successful elective percutaneous coronary intervention of a native artery. Am J Cardiol 2009;103:639-45.
91 -Omland T, de Lemos JA, Sabatine MS, et al. A sensitive cardiac troponin T assay in stable coronary artery disease. New Engl J Med 2009;361:2538-47.
92 -Klein Gunnwiek JMT, van der Hoeven JG. Cardiac troponin elevations among critically ill patients. Curr Opin Crit Care 2004;10:342-6.
93 -Lim Wendy, Cook DJ, Griffith LE, Crowther MA, Devereaux PJ. Elevated cardiac troponin levels in critically ill patients: prevalence, incidence and outcome. Am J Crit Care 2006; 15:280-8.
94 -Lim Wendy, Qushmaq I, Devereaux PJ, et al. Elevated cardiac troponin measurements in critically ill patients. Arch Intern Med 2006;166:2446-54.
95 -King DA, Codish S, Novack V, Barski L, Almog Y. The role of cardiac troponin I as a prognosticator in critically ill medical patients: a prospective observational cohort study. Critical Care 2005;9:R390-5.
96 -Stein R, Gupta B, Agarwal S, et al. Prognostic implications of normal (< 0.010ng/ml) and borderline (0.10 – 1.49ng/ml) troponin elevationlevelsin criticallyillpatients without acute coronary syndrome. Am J Cardiol 2008;102:509-12.
97 -Ammann P, Maggiorini M, Bertel O, et al. Troponin as a risk factor for mortality in critically ill patients without acute coronary syndromes. J Am Coll Cardiol 2003;41: 2004-9.
98 -Srivathsan K, Showalter J, Wilkens J et al. Cardiovascular outcome in hospitalized patients with minimal troponin I elevation and normalcreatine phosphokinase. Int J Cardiol 2004;97:221-4.
99 -Mehta NJ, Khan IA, Gupta V, et al. Cardiac troponin I predicts myocardial dysfunction and adverse outcome in septic shock. Int J Cardiol 2004;95:13-7.
100 -Purcell JB, Patel M, Khera Amit, et al. Relation of troponin elevation to outcomein patients withinfective endocarditis. Am J Cardiol 2008;101:1479-81.
101 -Pruszczyk P, Szule M, Torbicki A. Troponin levels in acute pulmonary embolism. Chest 2002;122:2264.
102 -Horwich TB, PatelJ, MacLellan WR, et al. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation 2003;108:833-8.
103 -Fonarow GC, Peacock WF, Horwich TB, et al. Usefulness of B-type natriuretic peptide and cardiac troponin levels to predict in-hospital mortality from ADHERE. Am J Cardiol 2008;101:231-7.
104 -Yin WH, Chen JW, Feng AN, Lin SJ, Young S. Multi-marker approach to risk stratification among patients with advanced chronic heart failure. Clin Cardiol 2007; 30:397-402.
105 -Miller WL, Hartman KA, Burritt MF, Burnett JC, Jaffe AS. Troponin B-Type natriuretic peptides and outcomes in severe heart failure: differences between ischemic and dilated cardiomyopathies. Clin Cardiol 2007;30:245-50.
106 -You JJ,Austin PC,Alter DA, Ko DT,Tu JV. Relation between cardiac troponin I and mortality in acute decompensated heart failure. Am Heart J 2007;153:462-70.
107 -Naidech AM, Kreiter KT, Janjua N, et al. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 2005;112:2851-6.
108 -Tanabe M, Crago EA, Suffoletto MS, et al. Relation of elevation in cardiac troponin I to clinical severity, cardiac dysfunction, and pulmonary congestion in patients with subarachnoid hemorrhage. Am J Cardiol 2008;102:1545- 50.
109 -Sandhu R, Aronow WS, Rajdev A, et al. Relation of cardiac troponin I levels with in-hospital mortality in patients with ischemic stroke, intracerebral hemorrhage, and subarachnoid hemorrhage. Am J Cardiol 2008;102:632-4.
110 -Gaudino M, Alessandrini F, Glieca F, et al. Survival after aortic valve replacement for aortic stenosis: does left ventricular mass regression have a clinical correlate? Eur Heart J 2005;26:51-7.
111 -Kupari M, Eriksson S, Turto H, Lommi J, Pettersson K. Leakage of cardiac troponin I in aortic valve stenosis. J Intern Med 2005;258:231-7.
112 -Adabag AS, Rector T, Mithani S, et al. Prognostic significance of elevated cardiac troponin I after heart surgery. Ann Thorac Surg 2007;83:1744-50.
113 -Steams JD, Dávilla-Román VG, BarzilaiB, et al. Prognostic value of troponin I levels for predicting adverse cardiovascular outcomes in postmenopausal women undergoing cardiac surgery. Anesth Analg 2009;108:719-26.
114 -Nesher N, Alghamdi AA, Singh SK, et al. Troponin after cardiac surgery:Apredictor or a phenomenon?Ann Thorac Surg 2008;85:1348-54.
115 -Bolliger D, Seeberger MD, Buse GALL, et al. A preliminary report on the prognostic significance of preoperative brain natriuretic peptide and postoperative cardiac troponin in patients undergoing major vascular surgery. Anesth Analg 2009;108:1069-75.
116 -Mantha S, Foss J, Ellis JE, Roizen MF. Intense cardiac troponin surveillance forlong-term benefitsis cost-effective in patients undergoing open abdominal aortic surgery: a decision analysis model. Anesth Analg 2007;105:1346-56.
117 -Apple FS, Murakami MM, Pearce LA, Herzog CA. Predictive value of cardiac troponin I and T for subsequent death in end-stage renal disease. Circulation 2002;106: 2941-5.
118 -DeFilippi C, Wasserman S, Rosanio S, Tiblier E, Sperger H, Tocchi M, et al. Cardiac troponin T and C-reactive protein for predicting prognosis, coronary atherosclerosis, and cardiomiopathy in patients undergoing long-term hemodialysis. JAMA 2003;290:353-9.
119 -Melloni C, Alexander KP, Milford-Beland S, Newby K, et al. Prognostic value of troponin in patients with non-ST-segment elevation acute coronary syndromes and chronic kidney disease. Clin Cardiol 2008;31:125-9.
120 -Shan K, Lincoff AM, Young JB. Anthracycline-induced cardiotoxicity. Ann Intern Med 1996;125:47-58.
121 -Horacek JM, Pudil R, Tichy M, et al. Cardiac troponin I seems to be superior to cardiac troponin T in the early detection of cardiac injury associated with anthracycline treatment. Onkologie 2008;31:559-60.
122 -Göser S, Andrassy M, Buss SJ, Leuschner F, et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation 2006;114:1693-702.
123 -Baillard C, Boussarsar M, Fosse JP, et al. Cardiac troponin Iin patients with severe exacerbation of chronic obstructive pulmonary disease. Intensive Care Med 2003;29:584-9.
124 -Harvey MG, Hancox RJ. Elevation of cardiac troponins in exacerbation of chronic obstructive pulmonary disease. Emerg Med Australasia 2004;16:212-5.
125 -Brekke PH, Omland T, Holmedal SH, Smith P, Soyseth V. Troponin T elevation and long-term mortality after chronic obstructive pulmonary disease exacerbation. Eur Respir J 2008;31:563-70.
126 -Martins CS, Rodrigues MJ, Miranda VP, Nunes JP. Neth Prognostic value of cardiac troponin I in patients with COPD acute exacerbation. J Med 2009;67:341-9.
Correspondência:
Dr.ª Carla Sofia Martins
Serviço de Medicina Interna
Hospital de São João
Alameda Prof. Hernâni Monteiro
4200-319 Porto
e-mail: carlamrt@hotmail.com