








Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArquivos de Medicina
On-line version ISSN 2183-2447
Arq Med vol.23 no.4 Porto Aug. 2009
Gravidez e Doença de Wilson
Revisão de 3 Casos Clínicos
Maria Geraldina Castro*, Rita Sousa†, Susana Marta†, Jorge Braga†
*Serviço de Ginecologia/Obstetrícia da Maternidade Bissaya Barreto, Centro Hospitalar de Coimbra; †Serviço de Ginecologia/Obstetrícia, Hospital Geral de Santo António-Centro Hospitalar do Porto
A doença de Wilson é uma doença hereditária autossómica recessiva que afecta o gene ATP7B, localizado no cromossoma 13. Caracteriza-se por uma alteração do transporte transmembranar do cobre, acumulando-se este no fígado e noutros órgãos, principalmente cérebro, rins e córnea. A prevalência mundial é de aproximadamente 30 casos por milhão de habitantes. Cerca de 60 a 70% dos casos são diagnosticados entre os 8 e os 20 anos de idade. Clinicamente, apresenta-se como doença hepática, neurológica ou psiquiátrica. O seu diagnóstico é baseado num alto índice de suspeição e na combinação de achados clínicos e laboratoriais. O exame gold standard para o diagnóstico é a biópsia hepática com doseamento do cobre. O tratamento consiste na terapêutica farmacológica permanente ou no transplante hepático. Existem actualmente três fármacos disponíveis: a penicilamina e a trientina (agentes quelantes) e o zinco. O tetratiomolibdato é um agente quelante ainda em investigação. A capacidade reprodutiva das mulheres com doença de Wilson tem melhorado com o aumento da eficácia destas terapêuticas e, como tal, a gravidez torna-se mais frequente. Esta parece não afectar o curso da doença e o principal problema que se coloca é a escolha da terapêutica mais adequada durante a gestação, dado que a segurança destes fármacos na gravidez não está garantida. Os autores descrevem 3 casos de grávidas com doença de Wilson, vigiadas na sua instituição, e discutem a controvérsia em torno do uso destes fármacos no tratamento da doença durante a gravidez e amamentação.
Palavras-chave: doença de Wilson; gravidez; disfunção hepática; trientina; penicilamina; zinco.
Pregnancy and Wilson Disease
Wilson disease is an autosomal recessive inherited disorder, with an affected gene (ATP7B) located at chromosome 13. It has been shown to affect the copper transport leading to a hepatic accumulation and deposition in other organs, specially brains, kidneys and cornea. The worldwide prevalence of Wilson's disease is around 30 per million. Around 60-70% of all cases are diagnosed between the ages of 8 to 20 years old.
Clinically the condition may present with hepatic, neurologic or psychiatric disease. The diagnosis is based on a high level of suspicion and on the combination of both clinical and laboratorial findings. The gold standard exam for Wilson's disease diagnosis is the liver biopsy with determination of hepatic copper levels. The treatment is lifelong pharmacological therapy or hepatic transplant. Currently there are three available drugs, penicillamine and trientine (chelating agents), and zinc. Tetrathiomolybdate is another chelating agent still under evaluation. The reproductive ability of women with Wilson disease has improved with the increasing efficiency of these therapies, and therefore pregnancy is becoming more common. It does not seem that the course of the disease is affected by pregnancy, but the problem remains on what is the most adequate therapy during gestation, as the safety of these drugs can not be assured during this period. The authors describe 3 cases of Wilson disease and pregnancy, followed at their centre and discuss the controversy around the use of these drugs for the treatment of the disease during pregnancy and lactation.
Key-words: Wilson disease; pregnancy; hepatic dysfunction; trientine; penicillamine; zinc.
INTRODUÇÃO
A doença de Wilson foi descrita pela primeira vez em 1912 por Kinnear Wilson como“degenerescência lenticular progressiva”, uma doença familiar, neurológica, fatal, acompanhada por doença hepática crónica e cirrose (1). Só décadas mais tarde foi descoberto o seu padrão hereditário autossómico recessivo e, em 1993, foi identificado o gene afectado nesta patologia, o ATP7B (2-5). Hoje em dia foram já descritas mais de 200 mutações neste gene, responsáveis pela doença. Este gene, localizado no braço longo do cromossoma 13, codifica uma ATPase transportadora de cobre, expressa essencialmente nos hepatócitos, actuando no transporte transmembranar de cobre. A diminuição da função desta proteína provoca uma redução da excreção hepatocelular do cobre no líquido biliar, resultando na acumulação hepática deste metal e lesão dos hepatócitos. O cobre é libertado na corrente sanguínea e deposita-se noutros órgãos, principalmente cérebro, rins e córnea. A alteração funcional da proteína ATP7B conduz também a uma falha na ligação do cobre à ceruloplasmina, pelo que o fígado produz e secreta apoceruloplasmina (ceruloplasmina sem cobre), com diminuição dos níveis séricos de ceruloplasmina (6).
EPIDEMIOLOGIA
A prevalência mundial da doença de Wilson é de cerca de 30 casos por milhão de habitantes, com aumento da incidência em áreas de consanguinidade (6). A taxa de heterozigotos para a doença é de 1 caso por 100 habitantes, correspondendo a uma frequência do gene de 0,3 a 0,7% (7).
Apesar da doença poder manifestar-se em qualquer idade, predomina nos jovens, com 60 a 70% dos casos diagnosticados entre os 8 e os 20 anos, sendo mais rara antes dos 3 e após os 40 anos (8).
Apresenta-se clinicamente como doença hepática, doença neurológica progressiva ou doença psiquiátrica. O envolvimento hepático ocorre, em geral, antes dos 20 anos, com o pico entre os 10 e 13 anos, enquanto que o atingimento neurológico tende a surgir após os 20 anos de idade (9).
CLÍNICA
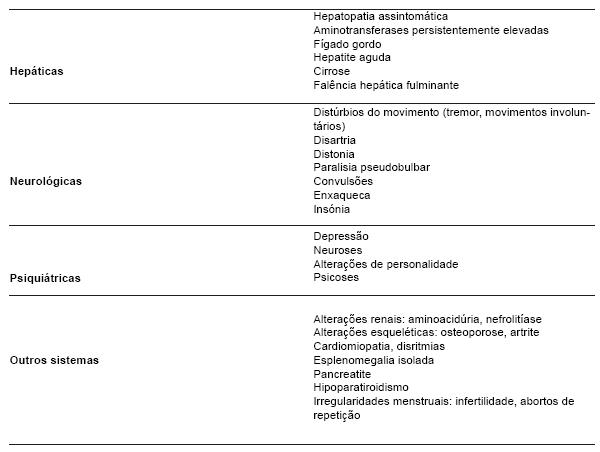
A doença de Wilson é progressiva e fatal se não for diagnosticada e tratada precocemente. Existem três principais formas de manifestação de doença hepática: hepatite crónica activa, cirrose e hepatite fulminante. A cirrose é a forma de apresentação inicial mais comum. A falência hepática fulminante, a forma de apresentação mais rara, surge acompanhada de anemia hemolítica Coombs negativa e falência renal aguda, que pode culminar com a morte se não for efectuado um transplante hepático emergente (6).
As manifestações neurológicas podem variar desde alterações motoras como tremor assimétrico (a mais comum), distonia, disartria, espasticidade, perda da coordenação motora, a alterações psiquiátricas, nomeadamente de comportamento, afectivas, esquizofrénicas e cognitivas.
O atingimento oftalmológico traduz-se pelo aparecimento de anéis de Kayser-Fleischer, formados pela deposição de cobre na membrana de Descemet da córnea. Estes observam-se em mais de 90% dos doentes com doença de Wilson sintomática e estão quase invariavelmente presentes nos doentes com manifestações neurológicas. No entanto, apesar de úteis no diagnóstico, não são considerados patognomónicos da doença, excepto quando acompanhados de manifestações neurológicas.
Clinicamente, a doença de Wilson pode ter também manifestações músculo-esqueléticas, hematológicas, renais, cardíacas, hormonais (Tabela 1).
Tabela 1 - Manifestações clínicas da doença de Wilson.
DIAGNÓSTICO
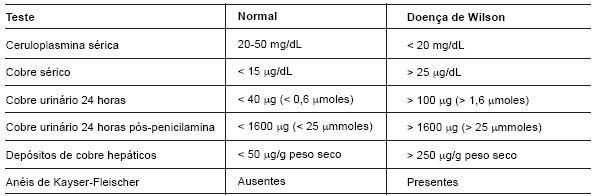
O diagnóstico da doença de Wilson baseia-se num alto índice de suspeição e na combinação de achados clínicos e laboratoriais. Os doentes sintomáticos têm invariavelmente valores de cobre urinário nas 24 horas superiores a 100 µg (normal: 20-50 µg). Os doentes com sintomatologia neurológica ou psiquiátrica apresentam anéis de Kaiser-Fleischer. Estes estão ausentes em 50% dos indivíduos com atingimento hepático como forma de apresentação ou pré-sintomáticos. Os níveis séricos de ceruloplasmina estão diminuídos em 80 a 90% dos doentes, no entanto, são normais em 10 a 20% e os indivíduos heterozigotos para o gene também apresentam valores baixos de ceruloplasmina sérica. O exame gold standard para diagnóstico da doença de Wilson é a biópsia hepática com doseamento de depósitos de cobre, cujos valores são superiores a 250 µg/g de peso seco de tecido (normal: 20 a 50 µg). Os heterozigotos têm uma elevação ligeira deste valor, mas nunca acima dos 200 µg (Tabela 2) (10).
Tabela 2 - Diagnóstico da doença de Wilson.
Actualmente, o diagnóstico genético ainda não está disponível, dado o elevado número de mutações do gene que se manifestam com o fenótipo desta doença. No entanto, quando é diagnosticado um caso índex numa família, a sua mutação é identificada e rastreada nos familiares em primeiro grau do probando (6).
TRATAMENTO
O tratamento da doença de Wilson passa pela terapêutica farmacológica durante toda a vida e pelo transplante hepático, que corrige o defeito hepático subjacente, reservado para os casos severos (hepatite fulminante) ou resistentes. A escolha dos fármacos e suas dosagens vai depender da apresentação clínica da doença. Nos doentes sintomáticos ou com doença activa a terapêutica recomendada é com agentes quelantes (penicilamina e trientina), uma vez que é necessário remover o cobre acumulado nos tecidos. A penicilamina é o fármaco utilizado com mais experiência, mas, actualmente, a trientina começa a ser usada com mais frequência, principalmente nos casos de doença neurológica descompensada (6). A terapêutica combinada com agentes quelantes e zinco, baseada na associação dos mecanismos de acção de bloqueio da absorção do cobre e eliminação do seu excesso, está ainda em estudo e a sua eficácia por determinar.
Uma vez estabilizada a sintomatologia e as alterações bioquímicas, dois a seis meses após a terapêutica inicial, podem ser usadas doses de manutenção, para prevenção da reacumulação do cobre. Os doentes assintomáticos no momento do diagnóstico podem ser tratados desde o início com doses de manutenção de qualquer um dos fármacos. À terapêutica médica deve ser acrescida uma dieta pobre em cobre, nomeadamente com evicção da ingestão de mariscos, fígado, chocolate e cogumelos.
Actualmente existem três fármacos disponíveis no mercado e um outro sob investigação.
O zinco aumenta os níveis de metalotionina, uma proteína intestinal com alta afinidade para o cobre, que se liga a este no enterócito duodenal. Desta forma, a absorção do cobre na circulação é reduzida e este é eliminado no turnover celular normal da célula. Além disto, o zinco induz a metalotionina nos hepatócitos, produzindo complexos não tóxicos no fígado e reduzindo os efeitos nocivos do cobre livre. O zinco está indicado como terapêutica de primeira linha nos doentes pré-sintomáticos ou assintomáticos, durante a terapêutica de manutenção e nos doentes com envolvimento neuro-psiquiátrico, uma vez que raramente provoca deterioração neurológica. É um fármaco bem tolerado, mas em 10 a 15% dos casos provoca irritação gástrica, que pode ser resolvida alterando a formulação (para acetato, sulfato ou gluconato) ou a hora de administração (11). No entanto, como os alimentos interferem na sua absorção, deve ser ingerido fora das refeições. A dose terapêutica é de 150 a 200 mg por dia, distribuída por três doses (Tabela 3).
Tabela 3 - Terapêutica da Doença de Wilson.
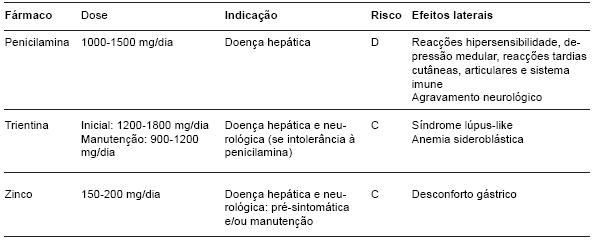
A penicilamina mobiliza o cobre através da sua acção quelante, formando complexos com este metal, que são depois excretados na urina. Além disto, também induz a metalotionina a nível hepático, promovendo o sequestro do cobre livre intracelular. A dose inicial é de 1000 a 1500 mg por dia, dividida por duas a quatro doses, 1 hora antes ou 2 horas após as refeições. Pode ser iniciada em doses mais baixas, 250 a 500 mg/dia, aumentando progressivamente, para uma melhor tolerabilidade. Este fármaco induz uma deficiência de vitamina B6, pelo que deve associar-se 25 mg de piridoxina diariamente. Os doentes hepáticos podem notar uma melhoria clínica após 6 a 8 semanas de tratamento, mas pode demorar 6 a 12 meses (12). Os efeitos adversos podem ocorrer em 10 a 20% dos doentes (13), e podem ser precoces ou tardios. Os primeiros, que ocorrem até às três semanas de tratamento, são as reacções de hipersensibilidade (febre, rash cutâneo, linfadenopatia e proteinúria) e a depressão medular com anemia aplástica, neutropenia e trombocitopenia, que devem levar à interrupção do fármaco. Durante o tratamento está recomendada uma monitorização regular do hemograma e da proteinúra devido aos efeitos adversos descritos. Os efeitos laterais tardios podem ocorrer anos após o uso da penicilamina e incluem atingimento cutâneo (alterações degenerativas, elastosis perforans serpiginosa) ou alterações imunológicas (reacções lupus-like, miastenia gravis, síndrome de Goodpasture). Uma deterioração neurológica inicial, descrita em 20 a 50% dos doentes com apresentação neurológica, pode surgir na terapêutica com penicilamina, que deve ser imediatamente substituída.
A trientina, tal como a penicilamina, é um quelante de cobre, que promove a sua excreção urinária e, como tal, pode ser usada como alternativa à penicilamina. Parece ter um efeito quelante um pouco mais fraco e provocar menos efeitos laterais. A dose inicial deve ser de 1200 a 1800 mg/dia, dividida em duas a três doses diárias e a dose de manutenção de 900 a 1200 mg/dia, ingerida fora das refeições. Os efeitos adversos descritos são a pancitopenia, raramente, e a anemia sideroblástica com siderose hepática, nos casos de deficiência de cobre por terapêutica excessiva. Reacções de hipersensibilidade e alterações renais não foram descritas. A frequência de deterioração neurológica é inferior à da penicilamina (inferior 20%).
O tetratiomolibdato de amónio é um fármaco ainda sob investigação, que apresenta dois mecanismos de acção. Quando tomado às refeições, liga-se ao cobre no lúmen intestinal, impedindo a sua absorção. Fora das refeições é absorvido e promove a formação de complexos de cobre e albumina, que são depois metabolizados no fígado e excretados na bílis. Este fármaco foi proposto para o tratamento inicial de doentes com apresentação neurológica da doença de Wilson (14). Estes doentes parecem apresentar deterioração neurológica mais raramente e cerca de 15% apresentam efeitos laterais moderados, tais como, toxicidade medular e elevação das enzimas hepáticas, ambas transitórias e reversíveis com a suspensão da droga.
Quando o tratamento é iniciado, deve ser realizada uma vigilância apropriada da parte neurológica e hepática, com exame físico dirigido e testes bioquímicos da função hepática. Durante o uso de quelantes, a excreção do cobre urinário durante 24 horas deve ser monitorizada (2 vezes no primeiro ano, depois anualmente), esperando-se um valor de 3 a 8 µmol (200-500 µg), indicativo de uma correcta adesão à terapêutica. No caso do uso de zinco, este valor deverá ser inferior a 1,2 µmol (75 µg). Durante todos os tratamentos devem ser medidos os níveis de ceruloplasmina livre e de cobre sérico, que permitem avaliar a eficácia terapêutica.
O prognóstico dos doentes de Wilson com atingimento hepático que aderem ao tratamento é excelente, mesmo quando a cirrose está presente no momento do diagnóstico (6).
Com a eficácia crescente destas terapêuticas também a capacidade reprodutiva das mulheres com doença de Wilson foi melhorada e, como tal, a gravidez é mais frequente. Esta parece não afectar o curso da doença e o principal problema que se coloca é qual a terapêutica mais adequada, uma vez que a segurança destes fármacos na gravidez não esta garantida (Tabela 3).
Os autores descrevem 3 casos de doença de Wilson e gravidez, vigiada na sua instituição, e discutem as controvérsias do uso dos medicamentos para o tratamento da doença de Wilson durante a gravidez e lactação.
CASOS CLÍNICOS
Caso clínico nº 1
Mulher de 39 anos, primigesta, com doença de Wilson diagnosticada há 1 ano, na sequência de investigação familiar por diagnóstico da mesma patologia num irmão. Apresentava hepatite crónica na biópsia hepática, cobre hepático elevado (386,47 µg/g peso seco), anéis de Kayser-Fleischer, ceruloplasmina baixa (9 mg/dL), cobre sérico (7,3 µmol/l) e urinário (2,142 µmol/24 horas) elevados, pelo que iniciou terapêutica com trientina. Efectuou estudo genético, tendo sido identificadas duas mutações do gene ATP7B, também encontradas no irmão. Um ano depois ficou grávida e iniciou vigilância pré-natal às 15 semanas de gestação, estando medicada nessa fase com 1000 mg de trientina por dia, que manteve até ao final da gravidez.
Foi efectuada vigilância obstétrica quinzenal até às 27 semanas e depois semanal, até ao fim da gestação. Realizou amniocentese às 20 semanas por idade materna avançada, cujo resultado foi normal (46,XY). Detectada brida amniótica na ecografia das 21 semanas, que persistiu até ao final da gestação, sem qualquer complicação.
Durante a gravidez não surgiu sintomatologia de novo relativamente à doença de Wilson. Analiticamente, apresentava ceruloplasmina baixa (15 mg/dL), cobre sérico normal, cobre urinário adequado para doentes em tratamento com agente quelante (5,3 µmol/24h) e função hepática normal.
Às 37 semanas realizou-se uma cesariana por rotura prematura de membranas e apresentação pélvica em primípara, com extracção de recém-nascido do sexo masculino, 2930 g de peso e Índice de Apgar (IA) 9/10. O puerpério decorreu sem incidentes, mantendo a mesma dose de trientina e com aleitamento materno.
Caso clínico nº 2
Mulher de 29 anos, primigesta, com doença de Wilson diagnosticada aos 16 anos, após episódio de falência hepática fulminante com hemólise severa, encefalopatia hepática e ascite, que reverteu com terapêutica médica. Apresentava anéis de Kayser-Fleischer, ceruloplasmina sérica baixa, cobre urinário e hepático elevados. Inicialmente tratada com penicilamina, mas, após quadro de febre e trombocitopenia, esta foi suspensa e iniciada trientina (750 mg/dia), com boa tolerância e sem efeitos laterais.
Iniciou vigilância pré-natal às 14 semanas de gestação, altura em que se encontrava medicada com trientina (750 mg/dia) e assintomática. Durante toda a gravidez apresentou ceruloplasmina baixa (inferior ao limite de detecção), cobre urinário adequado para doente em tratamento (5,33 µmol/dia) e função hepática normal. No terceiro trimestre da gravidez a dose de trientina foi reduzida para 500 mg/dia.
A gestação cursou sem complicações, com vigilância obstétrica quinzenal até às 30 semanas e semanal até às 36 semanas, altura em que ocorreu rotura prematura pré-termo de membranas (RPPM). Foi efectuada cesariana por falha de indução, tendo sido extraído um recém-nascido saudável, do sexo masculino, 3400 g de peso e IA 9/10. O puerpério decorreu sem complicações e a mãe amamentou a criança.
Caso clínico nº 3
Mulher de 35 anos, primigesta, com doença de Wilson diagnosticada aos 31 anos, após investigação familiar por diagnóstico da doença numa irmã. Encontrava-se assintomática, com ceruloplasmina sérica baixa (inferior ao limite de detecção), cobre urinário elevado (5,9 µmol/dia), cobre sérico e função hepática normais e biópsia hepática com infiltrado inflamatório e cobre hepático elevado. Iniciou terapêutica com penicilamina (900 mg/dia) e piridoxina (20 mg/dia). Efectuou consulta préconcepcional aos 32 anos. Iniciou vigilância pré-natal às 6 semanas de gestação e manteve terapêutica com 900 mg/dia de penicilamina até ao parto. Foi diagnosticada diabetes gestacional às 29 semanas, que foi controlada com dieta. A gravidez decorreu sem outras complicações. Os parâmetros clínicos e analíticos da doença de Wilson também se mantiveram estáveis.
Realizou-se cesariana às 38 semanas por apresentação pélvica em primípara, com extracção de um recém-nascido saudável, do sexo masculino, 3450 g de peso, IA 9/10. Puerpério sem complicações, com aleitamento materno.
DISCUSSÃO
Previamente à introdução da penicilamina, as gestações nas mulheres com doença de Wilson eram infrequentes, devido à redução da fertilidade provocada pela doença. Actualmente, com o diagnóstico e início precoce da terapêutica, as taxas de fertilidade melhoraram (15). Desta forma, a gravidez na mulher com doença de Wilson apresenta-se como um desafio. O objectivo da terapêutica na grávida será manter o controlo adequado da doença, evitar a teratogenicidade no feto e prevenir a interferência na cicatrização da ferida operatória no caso de cesariana.
Brewer e col. (11) relatam uma série de 19 mulheres tratadas com acetato de zinco durante 26 gestações, das quais resultaram 24 crianças normais, uma com defeito cardíaco congénito e uma com microcefalia. Defendem, por isso, que o zinco é o agente de escolha durante a gravidez, devido à sua segurança para o feto e que deverá ser mantido na dose de 50 mg, 3 vezes ao dia. Desconhecem-se efeitos teratogénicos em animais e humanos com este fármaco.
Scheinberg e Sternlieb (16) relatam uma série de 18 mulheres tratadas com penicilamina durante 29 gestações, das quais resultaram 29 crianças normais.
Sternlieb (17) faz uma revisão de casos publicados de grávidas com doença de Wilson, tratadas com agentes quelantes. Recolheu 111 casos de mulheres tratadas com penicilamina, durante 153 gestações, das quais resultaram 144 recém-nascidos normais, 2 abortos terapêuticos (um caso de hipertensão portal e varizes esofágicas e outro caso de espinha bífida), 2 abortos espontâneos e 3 recém-nascidos pré-termo. Foi descrito um caso de manosidose, um de fenda do palato e uma morte fetal in útero. Reuniu 17 casos de mulheres tratadas com trientina, durante 22 gestações, das quais resultaram 19 recém-nascidos normais, tendo-se procedido a um aborto terapêutico e um dos fetos apresentava isocromossoma X.
A segurança do uso da trientina durante a gravidez não está bem definida, uma vez que a sua experiência é mais limitada. Estudos em animais sugerem uma potencial teratogenicidade deste fármaco. A penicilamina pode ter efeito teratogénico em animais e humanos, tais como síndrome de cútis laxa, micrognatia (18) e foi já descrito um caso de embriopatia severa por penicilamina (19). No entanto, pensa-se que os efeitos teratogénicos dos agentes quelantes não se verificam nas doentes de Wilson, uma vez que as doses necessárias nesta patologia são inferiores às dos doentes com cistinúria (17).
A American Association for the Study of Liver Diseases sugere que o tratamento deve ser mantido durante a gestação e a dose deve ser reduzida em 25 a 50% da dose prégestacional, particularmente no terceiro trimestre, para permitir uma melhor cicatrização da ferida operatória, no caso de ser necessária uma cesariana (6). A interrupção da terapêutica associa-se a um alto risco de episódios hemolíticos com insuficiência hepática, falência hepática fulminante e morte materna (20,21). Portanto, parece que o risco de interromper a terapêutica é substancialmente maior do que o risco de teratogenicidade.
Também durante o aleitamento materno a terapêutica da doença de Wilson deverá ser mantida, pois não têm sido descritos efeitos nocivos para o bebé, apesar das concentrações de cobre e zinco noleite materno poderem estar reduzidas (17).
Os três casos descritos neste artigo vêm corroborar a importância da manutenção da terapêutica da doença de Wilson. Não ocorreu qualquer agravamento materno da doença nem nenhuma complicação na gravidez. De qualquer forma, seria importante a realização de estudos prospectivos para comparar a eficácia e segurança dos fármacos disponíveis para o tratamento da doença de Wilson, durante a gravidez.
REFERÊNCIAS
1-Wilson SAK. Progressivelenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912;34:295-507.
[ Links ]2 -Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 1993;5:327-37.
3 -Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 1993;5:344-50.
4 -Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet 1993;5:338-43.
5 -Yamaguchi Y, Heiny RE, Gitlin JD. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun 1993; 197:271-7.
6 -Roberts EA, Scilsky ML.AASLD guideline: a practice guideline on Wilson disease.Hepatology 2003;37:1475-88.
7 -Shar R et al. Wilson disease. Emedicine (serialonline) 2007 May. Disponível em: URL: http://www.emedicine.com
8 -Sternlieb I. Wilson’s disease. Clin Liver Dis 2000;4:229-39.
9 -Hortêncio APB, Júnior CAA, Lima JMC, Moreira DMQM, Moreira JO. Doença de Wilson e gravidez. Relato de caso. Rev Bras Ginecol Obstet 2001;23:329-32.
10 -Brewer GJ. Wilson disease. Curr Treat Options in Neurology 2000;2:193-204.
11 -Brewer GJ, Johnson VD, Dick RD, Fink JK, Kluin KJ. Treatment of Wilson’s disease with zinc. XVII: treatment during pregnancy. Hepatology 2000;31:364-70.
12 -MediciV, Rossaro L. Wilson disease: a genetic but treatable liver disorder. The HCV Advocate Medical Writer’s Circle (serial online) 2006 Aug. Disponível em: URL: http://www.hvadvocate.org.
13 -Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson disease. Lancet 2007;369:397-408.
14 -Brewer GJ et al. Treatment of Wilson disease with ammonium tetrathiomolybdato: III. Initial therapy in a total of 55 neurologically affected patients and follow up with zinc therapy. Arch Neurol 2003;60:379-85.
15 -Nunns D, Hawthorne B, Goulding P, Maresh M. Wilson’s disease in pregnancy. Eur J Obst & Gynecol Rep Biol 1995;62:141-3.
16 - Scheinberg IH, Sternlieb I. Pregnancy in penicillamine – treated patients with Wilson’s disease. N England J Med 1975;293:1300-2.
17 -Sternlieb I. Wilson’s disease and pregnancy. Hepatology 2000;31:531-532.
18 -Kaplan MM. Treatment of Wilson disease. UpToDate 2007.
19 -Pinter R, Hogge WA, McPherson E. Infant with severe penicillamine embryopathy born to a woman with Wilson disease. Am J Med Genet A 2004;128:294-8.
20 - 20. Stimono N, Ishibashi H, Ikematsu H et al. Fulminant hepatic failure during perinatal period in a pregnant woman with Wilson’s disease. Gastroenterol Jpn 1991;26:69-73.
21 -Das SK, Ray K. Wilson disease: an update. Nat Clin Pract Neurol 2006;2:482-93.
Correspondência:
Dr.ª Maria Geraldina Castro
Serviço de Ginecologia/Obstetrícia
Maternidade Bissaya Barreto
Centro Hospitalar de Coimbra
Quinta dos Vales
3041-801 Coimbra
e-mail: geraldinacastro@hotmail.com