








Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Ciências Agrárias
Print version ISSN 0871-018X
Rev. de Ciências Agrárias vol.35 no.2 Lisboa July 2012
Nova legislação aplicável à colocação de produtos fitofarmacêuticos no mercado - regulamento n.º 1107/2009
New legislation applicable to the placing on the market of plant protection products – regulation nº 1107/2009
Bento Pereira de Carvalho[1]
[1] MAMAOT - DGADR – Divisão de Homologação e de Avaliação Toxicológica, Ecotoxicológica, Ambiental e da Identidade dos Produtos Fitofarmacêuticos. E-mail: bcarvalho@dgadr.pt
RESUMO
O Regulamento (CE) n.º 1107/2009 do Parlamento Europeu e do Conselho, de 21 de Outubro de 2009, relativo à colocação de produtos fitofarmacêuticos (PF) no mercado, aplica-se desde 14 de Junho 2011. Tendo por objectivo garantir um elevado nível de protecção da saúde humana e animal e do ambiente, introduz novos requisitos na aprovação de substâncias activas e harmoniza requisitos para sinérgicos, protectores de fitotoxicidade e coformulantes. Pretende, ainda, eliminar, tanto quanto possível, os obstáculos ao comércio de produtos fitofarmacêuticos decorrentes da existência de diferentes procedimentos de autorização nos Estados-Membros, estabelecendo regras e prazos detalhados para a avaliação e decisão de pedidos de autorização de colocação no mercado de PF, incluindo normas relativas ao reconhecimento mútuo das autorizações e ao comércio paralelo, de modo a incrementar a livre circulação de tais produtos e a garantir a sua disponibilidade nos Estados-Membros, preservando simultaneamente a competitividade da agricultura da Comunidade.
Palavras-chave: Autorização, colocação no mercado, produto fitofarmacêutico, regulamento 1107/2009.
ABSTRACT
Regulation (EC) Nº 1107/2009 of the European Parliament and of the Council, of 21 October 2009, concerning the placing of plant protection products on the market, applies from 14 June 2011. Having as a purpose to ensure a high level of protection of both human and animal health and the environment, it introduces new requirements for the approval of active substances and harmonizes requirements for synergists, safeners and co-formulants. It aims also to eliminate as much as possible the barriers to the trade of plant protection products originating from the existence of different authorisation processes in the Member-States, establishing rules and detailed deadlines for the evaluation and decision of applications for placing on the market of plant protection products, including rules for the mutual recognition of authorisations and for parallel trade, to enhance the free movement of such goods and ensure their availability in the Member-States, preserving at the same time the competitiveness of Community agriculture.
Keywords: Authorization, placing on the market, plant protection products, regulation 1107/2009.
INTRODUÇÃO
O Regulamento (CE) n.º 1107/2009 do Parlamento Europeu e do Conselho, de 21 de Outubro de 2009, relativo à colocação de produtos fitofarmacêuticos no mercado (adiante designado de Regulamento 1107/2009), publicado no JOUE L309 de 24 de Novembro de 2009, tendo entrado em vigor 20 dias após a sua publicação, é aplicável na sua plenitude desde 14 de Junho de 2011. O objectivo deste artigo é realizar uma panorâmica sobre as alterações introduzidas por este diploma na colocação no mercado de produtos fitofarmacêuticos, anteriormente regulada pela Directiva n.º 91/414/CEE, agora revogada. O Regulamento 1107/2009 constitui um novo marco na homologação de produtos fitofarmacêuticos a nível europeu, seja pelos novos requisitos e prazos que introduz, seja pela exigência de coordenação acrescida entre autoridades nos diferentes Estados-Membros (EM) ou pela harmonização da prestação de informação ao público. Clarifica e harmoniza regras e procedimentos, seja para a aprovação de substâncias activas a nível europeu, seja para a autorização de produtos fitofarmacêuticos a nível nacional. Introduz ainda, a prazo, a necessidade de avaliação de protectores de fitotoxicidade e sinérgicos, bem como de coformulantes e a exigência de autorização de adjuvantes de mistura extemporânea, figura legal que já vigorava em Portugal anteriormente.
O Regulamento 1107/2009 não carece de transposição para o direito nacional, sendo de aplicação directa em Portugal, de acordo com os tratados europeus em vigor.
OBJECTIVOS DO REGULAMENTO 1107/2009
Ao contrário da directiva que o antecedeu, o Regulamento 1107/2009 não define somente um conjunto mínimo de requisitos a cumprir para a aprovação de substâncias activas e autorização de produtos fitofarmacêuticos, estabelecendo medidas inviabilizadoras de regras específicas nacionais, para além de certos limites claramente delimitados, contribuindo para a harmonização de procedimentos e regras nos Estados-Membros. Fixa ainda prazos rígidos para a execução dos diversos procedimentos, prevendo inclusive medidas de contingência no caso de necessidade de pedidos de dados adicionais aos requerentes.
Entre outros, o Regulamento 1107/2009, expressa nos seus considerandos como objectivo, a garantia de um elevado nível de protecção da saúde humana e animal e ambiente, preservando a competitividade da agricultura europeia. Numa leitura mais cuidada, verifica-se que o legislador procurou assegurar, tanto quanto possível, a igualdade concorrencial da produção agrícola no acesso ao mercado de produtos fitofarmacêuticos e a aproximação possível a um mercado único de produtos fitofarmacêuticos, num domínio, onde a livre circulação de bens está restringida, devido à necessidade de um procedimento de autorização em cada EM e pretendem-se minimizar barreiras técnicas e comerciais, salvaguardando a segurança para o Homem e para o ambiente, face a condições específicas nos EM.
Para esse efeito, a Europa é dividida em três zonas geográficas e é introduzida a figura da avaliação zonal de um pedido de autorização por um EM Relator Zonal (EMRZ), que é comentada ou auditada pelos outros EM da zona, de modo a assegurar a confiança global no sistema introduzido. Posteriormente e em regra, a decisão tomada pelo EMRZ deve ser respeitada por todos os EM dessa zona. É ainda introduzida a figura do reconhecimento mútuo obrigatório de autorizações concedidas noutros EM da mesma zona, sujeita a condições e prazos rígidos, de modo a reduzir barreiras técnicas entre EM. Finalmente, é criada a obrigatoriedade da figura do comércio paralelo entre EM (que anteriormente era aplicação voluntária pelos EM, mas já regulado em Portugal), de modo a permitir à produção agrícola evitar distorções de mercado entre EM.
É ainda objectivo do regulamento o fomento da transparência do procedimento administrativo, através da introdução de requisitos mínimos de divulgação de informação técnico-científica e administrativa ao público e a terceiros que desejem entrar no mercado dos produtos fitofarmacêuticos, nomeadamente de modo a suprir a não repetição de estudos em vertebrados.
O regulamento pretende ainda a indução da partilha de recursos entre autoridades competentes nos EM e entre agentes no mercado, mas poder-se-á concluir que reforça igualmente a competitividade no mercado europeu de produtos fitofarmacêuticos, nomeadamente através do fomento da concorrência entre autoridades competentes dos EM de uma mesma zona, introduzida pela livre escolha do EMRZ por parte do requerente de uma autorização.
ÂMBITO E MECANISMO GERAL DE APROVAÇÃO DE SUBSTÂNCIAS ACTIVAS E DE AUTORIZAÇÃO DE PRODUTOS FITOFARMACÊUTICOS
A definição de produto fitofarmacêutico não se alterou do disposto na Directiva 91/414/CEE. O Regulamento 1107/2009 estipula ainda que um produto fitofarmacêutico apenas pode ser colocado no mercado ou utilizado se tiver sido autorizado no EM em questão (art.28.º). Assim, é clarificada uma norma da Directiva 91/414/CEE que considerava a utilização como uma forma de colocação no mercado. Em acréscimo o regulamento define claramente as situações em que é dispensada uma autorização de colocação no mercado (arts.28.º, 53.º, 54º):
• Produtos que contenham exclusivamente substâncias de base;
• Produção, armazenamento ou transporte de produtos fitofarmacêuticos destinados a ser utilizados noutro Estado-Membro ou à exportação (semelhante ao disposto na Directiva 91/414/CEE);
• No caso de existir uma autorização de comércio paralelo (art.52.º);
• Autorização para situações de emergência fitossanitária – 120 dias (art.53.º);
• Autorização para efeitos de experimentação (art.54.º).
Salienta-se que, para várias das situações referidas de derrogação à necessidade de obtenção de uma autorização de colocação no mercado, são necessários outros tipos de autorização específicos.
Para a obtenção de uma autorização de colocação no mercado de um produto fitofarmacêutico, é necessário que as substâncias activas, os protectores de fitotoxicidade e os agentes sinérgicos contidos na preparação estejam aprovados (art.29.º). Se estes forem de origem distinta daquela que suportou a aprovação ou, com alteração do processo ou da localização do fabrico, dever-se-á confirmar a sua equivalência (art.38.º). A preparação não poderá ainda conter coformulantes listados como inaceitáveis (Anexo III). Entre os requisitos elencados para autorização de um produto fitofarmacêutico, quanto à segurança para o Homem e para o ambiente, salienta-se a necessidade de se encontrarem estabelecidos os limites máximos de resíduos para os usos autorizados, harmonizados a nível europeu ao abrigo do Regulamento n.º 396/2005.
APROVAÇÃO DE SUBSTÂNCIAS ACTIVAS
Como novidade face à Directiva 91/414/CEE, o Regulamento 1107/2009 introduz o conceito de divisão das substâncias em vários tipos:
• Substâncias activas, que designaríamos de comuns (em oposição às seguintes);
• Substâncias activas de baixo risco (art.22.º);
• Substâncias de base (art.23.º);
• Substâncias activas candidatas para substituição (art.24.º).
Os critérios para aprovação como substância activa de baixo risco constam no capítulo 5 do Anexo II ao regulamento e os critérios que definem uma substância activa candidata para substituição são definidos no capítulo 4 do mesmo anexo. As substâncias de base são definidas como substâncias aprovadas a nível europeu para outros efeitos que não a protecção de plantas, mas para as quais possa existir um uso como produto fitofarmacêutico. A Comissão Europeia é responsável pela publicação de uma lista de candidatos para substituição e de substâncias de base, sendo as substâncias de baixo risco definidas como tal quando da sua aprovação ou renovação da mesma.
Os critérios gerais de aprovação das substâncias activas constam do Anexo II do Regulamento 1107/2009, sendo introduzidos critérios primários de aprovação das substâncias activas (critérios cut-off em jargão europeu) e a necessidade de demonstração de eficácia e inexistência de fitotoxicidade do produto fitofarmacêutico que suporta o pedido de provação, para os usos pedidos. É ainda definido o conceito da necessidade de pelo menos um uso representativo para pelo menos um produto fitofarmacêutico para suporte da aprovação da substância activa. A importância dos critérios primários de aprovação traduz-se no facto de se um destes critérios não se cumprir, a avaliação da substância activa termina nesse ponto, sendo prescindido o resto da avaliação da substância activa.
Contudo, o regulamento permite aprovar substâncias activas em derrogação dos critérios de aprovação 3.6.3, 3.6.4, 3.6.5 ou 3.8.2 do Anexo II, desde que para controlar um perigo fitossanitário grave que não possa ser combatido por outros meios disponíveis, incluindo métodos não químicos, mas só durante o prazo limitado necessário para controlar o perigo grave e não superior a cinco anos, sendo impostas medidas de redução dos riscos de exposição dos seres humanos e do ambiente. Esta derrogação não se aplica a substâncias activas classificadas como cancerígenas da categoria 1A ou da categoria 1B sem limiar ou tóxicas para a reprodução da categoria 1A.
Ao contrário da Directiva 91/414/CEE, são definidos procedimentos e prazos estritos para a aprovação, renovação e revisão das aprovações (art.7.º-21.º), sendo inclusivamente previstos prazos máximos para a submissão e para a avaliação de dados suplementares considerados necessários. É ainda prevista a colocação ao dispor do público do dossier sumário (art.10.º e 16.º) e da monografia (art.12.º), c/ exclusão de informação confidencial.
Enquanto a Directiva 91/414/CEE previa períodos de aprovação até 10 anos, agora são estipulados prazos distintos para aprovação e renovação, consoante o tipo de substância activa:
• Substâncias activas comuns – primeira aprovação 10 anos (art.5.º); renovação 15 anos (art.14.º);
• Substâncias activas de baixo risco – primeira aprovação 15 anos (art.22.º); renovação 15 anos (art.14.º);
• Substâncias de base – primeira aprovação por tempo ilimitado (art.23.º);
• Substâncias activas candidatas a substituição – primeira aprovação 7 anos; renovação 7 anos (art.24.º);
• Substâncias activas aprovadas em derrogação dos critérios de aprovação 3.6.3, 3.6.4, 3.6.5 ou 3.8.2 do Anexo II – primeira aprovação 5 anos (art.4.º); renovação 5 anos (art.14.º).
A relevância destes prazos distintos prende-se com a influência sobre a validade das autorizações de colocação no mercado de produtos fitofarmacêuticos que contenham os diferentes tipos de substância activa.
SINÉRGICOS E PROTECTORES DE FITOTOXICIDADE
Relativamente a sinérgicos e protectores de fitotoxicidade, será aprovado até 14 de Dezembro de 2014 um regulamento para definição dos requisitos de dados, dos procedimentos da sua avaliação e do programa de revisão das substâncias no mercado (art.26.º). Durante cinco anos após a aprovação do regulamento referido, podem ser autorizados produtos fitofarmacêuticos que contenham sinérgicos ou protectores de fitotoxicidade não aprovados, desde que incluídos no programa de revisão a estabelecer (art.81.º).
AUTORIZAÇÃO DE PRODUTOS FITOFARMACÊUTICOS
Tendo sido já abordados, em termos genéricos, os requisitos para a concessão de uma autorização de colocação no mercado de produtos fitofarmacêuticos (art.29.º), é de salientar a inovação introduzida pelo regulamento relativamente a este aspecto: ao abrigo da Directiva 91/414/CEE, encontravam-se estipulados critérios mínimos de segurança e de eficácia a serem cumpridos, ficando ao critério do EM a criação de regras ainda mais apertadas na transposição e aplicação daquela directiva. O Regulamento 1107/2009, contudo, não só replica esses critérios mínimos, como ainda acrescenta procedimentos e prazos a serem cumpridos para a avaliação e eventual concessão de autorizações.
São introduzidos os conceitos da avaliação zonal, de forma a obviar o reconhecimento de autorizações dadas por outros EM, e do reconhecimento mútuo e autorização de comércio paralelo obrigatórios. Apesar de diversas salvaguardas incluídas no diploma, o incumprimento daquelas obrigações por parte das autoridades competentes nos EM pode ser interpretado como um entrave ao mercado único e à livre circulação de produtos fitofarmacêuticos. As figuras da avaliação zonal e do reconhecimento mútuo interligados, não são nada mais do que um mecanismo que permite ao requerente de uma autorização realizar o pedido no EM que considere mais vantajoso, colocando em efectiva concorrência as autoridades competentes dos EM. O comércio paralelo, por seu turno, visa obstar às práticas comerciais das empresas detentoras de autorizações que originem distorções de mercado entre EM.
AVALIAÇÃO ZONAL (ARTS.33.º-37.º)
O Regulamento 1107/2009 divide politicamente a União Europeia em três zonas consideradas suficientemente uniformes, no conjunto das diversas áreas de avaliação de uma autorização, para que uma autorização concedida num EM dessa zona, possa ser dada noutro EM da mesma zona. As zonas são a zona Norte, Centro e Sul, encontrando-se Portugal na zona Sul, acompanhado de Espanha, França, Itália, Malta, Grécia, Chipre e Bulgária (Figura 1)
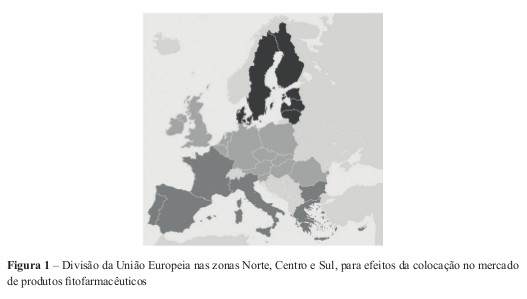
Todas as autorizações a conceder (incluindo extensões de utilização para produtos já autorizados) carecem de uma avaliação zonal a ser efectuada EMRZ, escolhido pelo requerente, excepto se outro EM aceitar realizar a avaliação. Contudo, o pedido de autorização deve ser efectuado em cada EM onde se pretenda a autorização, cabendo a estes EM coordenarem o seu trabalho de avaliação dos pedidos recebidos, sendo pressuposta a utilização uniforme dentro de cada zona.
Constituem excepção à divisão em três zonas as finalidades em estufa ou como tratamento pós-colheita, de locais de armazenamento vazios ou de sementes, situações em que deve ser proposto um único Estado-Membro, que avalia o pedido tendo em conta todas as zonas, sendo assumido que para aquelas situações não existirão diferenças entre as três zonas.
Durante a avaliação do EMRZ, os outros EM dever-se-ão abster de avaliar o pedido (ou cingir a sua avaliação aos requisitos específicos nacionais, exigíveis no caso de salvaguarda da saúde humana ou do ambiente face a situações particulares nacionais). Existindo pedidos efectuados em mais do que uma zona, os respectivos EMRZ dever-se-ão coordenar.
No final da sua avaliação o EMRZ produz um projecto de relatório de avaliação, de formato definido, que é colocado aos dispor dos outros EM da zona, para comentários, que são tidos em conta na versão final do relatório de avaliação, que consubstancia a autorização concedida.
O prazo concedido para relato e decisão do EMRZ é de 12 meses a contar da data do pedido, podendo este prazo ser dilatado por mais seis meses para submissão e avaliação de dados suplementares que sejam considerados necessários durante a avaliação. No caso de substâncias activas novas em avaliação e para um produto fitofarmacêutico e finalidades iguais aos do processo de aprovação dessa substância activa, o prazo para decisão reduz-se para seis meses, a contar da data de aprovação da substância activa.
Após decisão do EMRZ, os outros EM devem conceder ou recusar as autorizações pedidas, de acordo com as conclusões da avaliação efectuada pelo EMRZ, sendo o prazo de decisão limitado a 120 dias, podendo, contudo, ser impostos requisitos específicos e medidas de redução dos riscos, decorrentes de condições de utilização específicas nacionais. Pode ainda ser recusada a autorização, se, devido a circunstâncias ambientais ou agrícolas específicas, existir um risco inaceitável para a saúde humana ou animal ou para o ambiente, devendo nesse caso ser informada a Comissão Europeia desse facto.
RECONHECIMENTO MÚTUO (ARTS.40.º- 42.º)
O reconhecimento mútuo já existia de modo facultativo na Directiva 91/414/CEE, tendo sido implementado em Portugal desde Abril de 2009. O Regulamento 1107/2009 define procedimentos e requisitos harmonizados e instaura a figura da obrigatoriedade do reconhecimento mútuo.
O titular de uma autorização já concedida pode apresentar um pedido de autorização para o mesmo produto fitofarmacêutico, a mesma finalidade e com uma utilização de acordo com práticas agrícolas comparáveis noutro EM, se a autorização de referência tiver sido concedida:
• Na mesma zona;
• Em zona diferente, desde que não sirva para reconhecimentos mútuos subsequentes;
• Em zona diferente, para finalidades em estufa ou para tratamento pós-colheita, de locais de armazenamento vazios ou de sementes.
O procedimento é em tudo semelhante à decisão dos outros EM da zona, relativamente à decisão do EMRZ, no procedimento normal de autorização zonal, devendo a decisão ser tomada em 120 dias, sendo a autorização obrigatória, podendo ser impostos requisitos ou medidas de redução de riscos, devido a condições específicas nacionais, ou recusada, mas só em caso de risco inaceitável para a saúde humana ou animal ou para o ambiente, devendo nesse caso ser informada a Comissão Europeia desse facto.
VALIDADE E CANCELAMENTO DAS AUTORIZAÇÕES
A validade máxima de uma autorização é até 1 ano após caducidade da aprovação de qualquer das substâncias activas, protectores de fitotoxicidade e agentes sinérgicos contidos no produto (art.32.º).
No caso de cancelamento de uma autorização, pode ser dado um prazo para a distribuição e venda de seis meses a contar da data de cancelamento da autorização e um prazo suplementar de até 12 meses para a eliminação, o armazenamento e a utilização das existências (art.46.º).
CASOS ESPECIAIS
Produtos de baixo risco (art.47.º)
É criada a figura do produto fitofarmacêutico de baixo risco com uma definição, menos restritiva que a existente a nível nacional aplicável ao Decreto-Lei n.º 173/2005: enquanto anteriormente a lei nacional previa que um produto era de baixo risco, caso não exigisse classificação e não fossem exigidas medidas específicas de redução de riscos, o Regulamento 1107/2009 estipula que as preparações podem ter classificação, mas as substâncias activas contidas devem ser de baixo risco e que não podem estar contidas substâncias potencialmente perigosas. A grande distinção no regulamento para estes produtos é a exigência de tomada de decisão sobre qualquer pedido efectuado no prazo de 120 dias, com um prazo adicional de seis meses para submissão e avaliação de dados suplementares.
Sementes tratadas (art.49.º)
É estabelecida a livre circulação de sementes tratadas, desde que tratadas com produtos autorizados na UE, mas com indicação obrigatória do tratamento na rotulagem da semente. Esta medida já se encontrava implementada em Portugal, através do Decreto-Lei n.º 38/2009.
Produtos fitofarmacêuticos com candidatos para substituição (art.50.º e ponto 4 do Anexo II)
Sendo definidos os critérios de identificação de substâncias activas candidatas para substituição, o Regulamento 1107/2009 estabelece a obrigatoriedade da avaliação comparativa para os produtos que as contenham. Trata-se da verificação de existência de alternativas ao produto cuja autorização é pedida, que, cumulativamente, sejam significativamente mais seguras, não tenham desvantagens económicas ou práticas significativas e que seja assegurada a diversidade química das substâncias disponíveis ao utilizador, para a minimização da ocorrência de resistências. A autorização deve ser recusada ou restringida no caso de existência de alternativas satisfatórias e em número suficiente.
Avaliação comparativa pode ser aplicada voluntariamente pelos EM a outras substâncias activas, o que na prática poderá ser entendido como uma forma das autoridades competentes nos EM poderem restringir o reconhecimento mútuo obrigatório de autorizações noutros EM. A avaliação comparativa pode ainda ser adiada, para ganho prévio de experiência do uso do produto, sendo as autorizações nesse caso limitadas por cinco anos.
Extensões das autorizações para usos menores (art.51.º)
O Regulamento 1107/2009 institui a figura da extensão da autorização para usos menores, que era já uma prática corrente em alguns EM, nomeadamente em Portugal. Trata-se de conceder aquelas extensões a produtos já autorizados, mediante a apresentação de um conjunto limitado de dados. Podem ser pedidas pelo titular da autorização, o que é uma novidade, mas também por organismos oficiais ou científicos, organizações profissionais agrícolas e utilizadores profissionais. Como requisitos é exigido que o uso seja considerado menor no EM em causa (definição no art.3.º), que a extensão de autorização seja de interesse público, que existam dados de resíduos e, se necessário, relativos aos riscos para operador, trabalhador e transeunte. No caso destas extensões de autorização serem concedidas, estas podem ser inseridas no rótulo, com acordo do titular do produto. Em caso alternativo, é obrigatória a divulgação em publicação oficial ou em sítio web oficial. Em ambos os casos é necessário ser salientada uma declaração de exoneração de responsabilidade: a fitotoxicidade e a eficácia, são da responsabilidade do utilizador do produto naquelas utilizações.
É de referir ainda a aplicabilidade do reconhecimento mútuo, desde que o uso seja menor no EM onde é feito o pedido de extensão da autorização para uso menor.
Comércio paralelo (art.52.º)
Uma autorização de comércio paralelo, é a autorização de importar um produto já autorizado noutro EM, se aquele se encontrar igualmente autorizado no país onde se pretende a sua comercialização e utilização. Sendo uma prática não harmonizada anteriormente na UE, era uma figura jurídica já regulamentada em Portugal, através do Decreto-Lei n.º 22/2001, alterado pelo Decreto-Lei n.º 111/2007. Sendo uma disposição que exige coordenação entre autoridades competentes dos EM, são agora estipulados prazos de resposta a pedidos de informação de outros EM. De forma a harmonizar procedimentos e evitar distorções de mercado, além de serem estabelecidos requisitos e a obrigatoriedade da concessão da autorização, são definidos prazos para a decisão por parte da autoridade competente, sendo o objectivo que este procedimento seja muito ligeiro e célere. Os requisitos principais para esta autorização são que o produto a importar seja idêntico (ver definição) a um produto já autorizado no EM de introdução (produto de referência) e que as finalidades e condições de utilização sejam idênticas ao do produto de referência, podendo ser um subconjunto das mesmas.
Obrigações acessórias
Publicidade (art.66.º)
O Regulamento harmoniza igualmente a forma e o conteúdo da publicidade dos produtos fitofarmacêuticos: é interditada a publicidade de produtos não autorizados; a expressão baixo risco é interdita no rótulo, mas na publicidade dos produtos de baixo risco é permitida a expressão: autorizado como produto fitofarmacêutico de baixo risco nos termos do Regulamento (CE) n.º 1107/2009; A publicidade pode ser restringida em certos meios de comunicação, devendo todas as declarações ser tecnicamente justificáveis e sendo interditas representações visuais de práticas potencialmente perigosas; na publicidade ou no material de promoção deve ainda ser chamada a atenção para as frases de advertência adequadas e símbolos indicados na rotulagem.
Conservação de registos (art.67.º)
O Regulamento 1107/2009 assegura ainda a rastreabilidade do ciclo de vida do produto fitofarmacêutico, desde a sua produção à sua utilização. É tornada obrigatória a manutenção de registos durante:
• cinco anos – para a produção, importação, exportação, armazenamento e colocação no mercado;
• três anos para a utilização profissional, incluindo nome comercial do produto, data, dose, área e culturas.
Esta informação deve ser disponibilizada à autoridade competente, mediante pedido, que a poderá facultar a terceiros, se solicitada.
PRODUÇÃO DE EFEITOS E DIPLOMAS COMPLEMENTARES
Como já referido, o Regulamento 1107/2009 entrou em vigor em 14 de Dezembro de 2009, tendo a produção de efeitos iniciado a 14 de Junho de 2011. Nesta data entraram em aplicação, ainda, os seguintes regulamentos UE:
• Reg. 540/2011 – lista das substâncias activas aprovadas - actualizado sucessivamente com novas aprovações ou alterações às aprovações de substâncias activas;
• Reg. 544/2011 – requisitos de dados para substâncias activas;
• Reg. 545/2011 – requisitos de dados para produtos fitofarmacêuticos;
• Reg. 546/2011 – princípios uniformes de avaliação;
• Reg. 547/2011 – requisitos de rotulagem.