










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Clínica Geral
versão impressa ISSN 0870-7103
Rev Port Clin Geral vol.27 no.6 Lisboa nov. 2011
ESTUDOS ORIGINAIS
Estudo de microcitose numa população jovem adulta assintomática
Evaluation of microcytosis in an asymptomatic young adult population
Manuela Rebordão,* Manuel Silva*
*Licenciada em Ciências Farmacêuticas. Especialista em Análises Clínicas pela Ordem dos Farmacêuticos. Mestre em Imunologia Clínica pela Faculdade de Medicina da Universidade Beira Interior. Hospital Militar de Belém. Responsável de Hematologia do Serviço de Análises Clínicas
**Licenciado em Ciências Farmacêuticas. Tenente Coronel Farmacêutico. Especialista em Análises Clínicas pela Ordem dos Farmacêuticos. Hospital Militar de Belém. Chefe do Serviço de Análises Clínicas
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Objectivo: Quantificar e identificar as causas de microcitose de uma população jovem adulta assintomática.
População e Métodos: Foram estudados 2558 jovens candidatos a ingressar no Exército Português e previamente apurados nas provas físicas exigidas para o efeito. No grupo que apresentou volume globular médio inferior a 80fL (microcitose) foi efectuado doseamento de ferro, de ferritina e electroforese capilar de hemoglobina. Os casos foram agrupados de acordo com as possíveis causas de microcitose. Fez-se o levantamento do local de nascimento e residência. Foi aplicada estatística descritiva com o software SPSS.
Resultados: A prevalência de microcitose foi de 3,2% (N=81). Foram identificados quatro grupos. Grupo I (N=12, 15%), dez portadores de hemoglobina S (70% de Lisboa e Setúbal), um portador de hemoglobina C (de Lisboa) e um portador de hemoglobina Lepore (de Santarém). Grupo II (N=16, 20%), portadores heterozigóticos de β-talassémia, a maior parte proveniente de Faro. Grupo III (N=21, 26%), maioritariamente de Lisboa, com valores de ferritina inferiores a 15ng/mL e caracterizados por anemia ferropénica. Grupo IV (N=32, 40%), com valores normais de ferro, ferritina e electroforese de hemoglobina, suspeitos de serem portadores de α-talassémia, maioritariamente de Lisboa.
Conclusão: A avaliação da microcitose nesta população jovem adulta assintomática, feita a partir de uma metodologia simples e pouco dispendiosa, foi suficientemente robusta e capaz de identificar anomalias genéticas e carenciais. Contribuiu-se para um melhor conhecimento epidemiológico e atuação no sentido de se instituírem medidas preventivas e promotoras de saúde como sejam o aconselhamento e a orientação genética.
Palavras-chave: Índices de Eritrócitos; Talassémia; Hemoglobinopatias.
ABSTRACT
Aim: To identify the causes of microcytosis in an asymptomatic young adult population.
Population and Methods: Candidates for induction into the Portuguese army who had passed the induction physical examination were included in this study. For subjects with a mean corpuscular volume less than 80fL (microcytosis), analyses of serum iron, ferritin and hemoglobin electrophoresis were performed. Cases were grouped according to possible causes of microcytosis. Data on birthplace and place of residence were recorded. Descriptive statistics were analyzed with SPSS software.
Results: We studied 2558 subjects. The prevalence of microcytosis was 3,2% (N=81) in this population, we identified four diagnostic groups. Group I (N=12, 15%) included ten subjects with hemoglobin S (70% from Lisbon and Setúbal), a carrier of hemoglobin C (from Lisbon) and a carrier of hemoglobin Lepore (from Santarém). Group II (N=16, 20%) was composed of heterozygous carriers of β-thalassemia, mostly from Faro. Subjects in Group III (N=21, 26%) were mostly from Lisbon, with ferritin below 15ng/mL and were characterized by iron deficiency anemia. Group IV (N=32, 40%) included subjects with normal serum iron, ferritin and hemoglobin electrophoresis, suspected of being carriers of α-thalassemia, mostly from Lisbon.
Conclusion: The evaluation of microcytosis in an asymptomatic young adult population using simple and inexpensive methods was robust enough to identify subjects with genetic abnormalities and iron deficiency. This study contributes to a better epidemiologic understanding of this population. It enables the introduction of preventive and health-promoting measures such as counselling and genetic guidance.
Keywords: Erythrocyte Indices; Thalassemia; Hemoglobinopathies.
INTRODUÇÃO
A microcitose numa população jovem adulta aparentemente saudável é normalmente um achado laboratorial aquando da realização de um hemograma. É definida como um volume globular médio abaixo de 80fL, podendo estar ou não associada a anemia. As causas mais frequentes de microcitose são as anemias ferropénicas, as hemoglobinopatias e as talassémias.1
A anemia por carência de ferro tem várias etiologias, sendo as mais frequentes, na idade jovem adulta, uma dieta nutritiva deficiente, perdas de sangue por problemas gastrointestinais, perdas menstruais na mulher, diminuição da absorção por doença celíaca e infecção por Helicobacter pylori.2,3,4,5
As hemoglobinopatias e as talassémias são um conjunto de alterações hereditárias da molécula de hemoglobina que resultam de mutações nos genes responsáveis pela síntese das cadeias de globina da hemoglobina. Estas alterações podem-se traduzir em modificações estruturais da síntese das cadeias de globina (chamadas alterações qualitativas ou hemoglobinopatias) e/ou na diminuição ou ausência da síntese destas mesmas cadeias (alterações quantitativas ou talassémias).
De acordo com a Organização Mundial de Saúde (OMS), mundialmente, 270 milhões de pessoas (5% da população mundial) possuem genes que determinam hemoglobinas anormais. Estudos epidemiológicos mostram que 300 a 400 mil crianças nascidas vivas por ano apresentam estas patologias. A sua distribuição nas diferentes populações é muito variável, atingindo prevalências mais elevadas nas de origem mediterrânica, africana e oriental.6
Em Portugal os estudos apontam para uma prevalência de cerca de 1%, correspondendo a 5 a 10 novos casos diagnosticados por ano (0,45% portadores de β-talassémia e 0,32% portadores de drepanocitose). A maior prevalência encontra-se nos distritos de Lisboa, Setúbal e Faro, sendo também referenciados os distritos de Beja, Évora, Santarém e Leiria como prevalentes. O número crescente de doentes com drepanocitose, sobretudo na área da grande Lisboa, é reflexo da imigração da década de 70, proveniente das antigas colónias de África.7,8
Sabemos que a hemoglobina (Hb) no adulto é constituída por quatro cadeias de globina (2 alfa e 2 não alfa) e quatro grupos contendo ferro - heme. O tipo de hemoglobina depende da composição das cadeias de globina. Assim, no adulto existe cerca de 97% de HbA (α2β2), 2,2-3% de HbA2 (α2δ2) e 0,5-1% de Hb F(α2γ2).
A drepanocitose, ou anemia de células falciformes, surge quando há uma substituição anómala de um aminoácido na cadeia β da HbA (ácido-glutâmico por valina) resultando a chamada HbS. Esta hemoglobina em desoxigenação polimeriza, dando origem à distorção da forma do eritrócito e à sua rigidez, resultando uma anemia hemolítica crónica e episódios vaso-oclusivos.9 Estas manifestações clínicas passam despercebidas num portador heterozigótico, considerando-se assintomático.
Se houver uma outra substituição na mesma posição da cadeia β da HbA (ácido-glutâmico por lisina) surge-nos a HbC. Estas alterações na sequência dos aminoácidos alteram a carga eléctrica total da hemoglobina facilitando a sua detecção através da alteração da zona de migração na eletroforese.
A β-talassémia está presente quando num dos genes do par do cromossoma 11 existe uma mutação que leva a uma redução da síntese das cadeias β (β+/normal) ou a ausência da sua síntese (β0/normal), resultando uma anemia ligeira hipocrómica e microcítica (β-talassémia minor). Se os dois genes estiverem mutados mas com capacidade de produzirem algumas cadeias β (β+/β+) a anemia é moderada (β-talassémia intermédia), tornando-se severa e dependente de transfusões (β-talassémia major) quando não existe qualquer produção (β0/β0).10
A α-talassémia existe quando há uma mutação num ou mais dos quatro genes do cromossoma 16 que determinam a síntese das cadeias α. Assim, com três genes funcionantes (-α/αα) a diminuição da produção das cadeias α é reduzida, estando o VGM e o HGM no limite da normalidade (α-talassémia silenciosa). Microcitose, com ou sem anemia hipocrómica, é expressa quando só há dois genes activos (-α/-α) – traço de α-talassémia homozigótica α+, ou no traço de α-talassémia heterozigótica α0 (—/αα), tornando-se mais severa com um só gene (—/-α) - α-talassémia intermédia ou doença da HbH, e incompatível com a vida na ausência total da sua produção – hemoglobina de Bart.11
A Hb Lepore é formada por duas cadeias de globina alfa e duas outras que contêm um gene de fusão delta beta. Em heterozigotia expressam um fenótipo semelhante a uma talassémia minor.12
Nas talassémias o desequilíbrio na síntese das cadeias da globina conduz a uma redução da velocidade de síntese da hemoglobina e consequentemente uma hiperplasia eritróide compensatória com aumento da produção dos glóbulos vermelhos com tamanho e conteúdo reduzido.13
A identificação da causa de microcitose e a detecção precoce de portadores de hemoglobinopatias e talassémias numa população jovem adulta e em idade reprodutiva é de todo o interesse pois, se por um lado permite fazer um reconhecimento epidemiológico destas patologias, por outro, pode conduzir à instituição de medidas preventivas e promotoras de saúde, como seja um aconselhamento e orientação genética.
Objectivo: quantificar e identificar as causas de microcitose de uma população jovem adulta assintomática candidata a ingressar no Exército Português e previamente apurada nas provas físicas exigidas para o efeito.
MÉTODOS
Este estudo foi concebido no Laboratório de Análises Clínicas do Hospital Militar de Belém em cooperação com o Gabinete de Classificação e Selecção de Lisboa (GCSL), que selecciona candidatos a ingressarem no Exército Português, estendendo-se o limite territorial de Coimbra até ao Algarve. Todos os estudos analíticos do sangue e urina foram aqui executados, sendo o consentimento informado do estudo e a informação final da aptidão da responsabilidade do respectivo Gabinete.
A população estudada foi de 2558 jovens, entre Janeiro e Novembro de 2010, sendo 2102 do sexo masculino (82,1%) e 456 do sexo feminino (17,8%), com idade média de 20 anos (amplitude 17-29 anos e moda 19 anos). Toda a população foi estudada através de hemograma efectuado no equipamento Pentra ABX da Horiba. Todos os jovens que apresentavam um volume globular médio (VGM) inferior a 80fL foram considerados casos de microcitose. Nestes, foi executado, para além do preconizado na rotina (hemograma, bioquímica, marcadores virais), um esfregaço sanguíneo corado com May-Grunwald-Giemsa, doseamento do ferro e ferritina por quimioluminiscência no equipamento Architect da Abbott, e uma eletroforese de hemoglobina no equipamento Minicap da Phadia, cujo fundamento técnico é a electroforese capilar. O critério usado para definir o grupo da anemia ferropénica foi ter ferritina inferior ou igual a 15ng/mL na ausência de parâmetros inflamatórios.14 Foi feito o levantamento do local de nascimento e de residência de todos os casos.
O tratamento estatístico dos dados foi feito utilizando o programa SPSS.
RESULTADOS
Dos 2558 candidatos ao ingresso no Exército Português, 81 (3,2%) apresentavam microcitose com um VGM < 80 fL. O estudo do ferro, da ferritina e da eletroforese de hemoglobina permitiu separar estes jovens em quatro grupos (Figura 1).
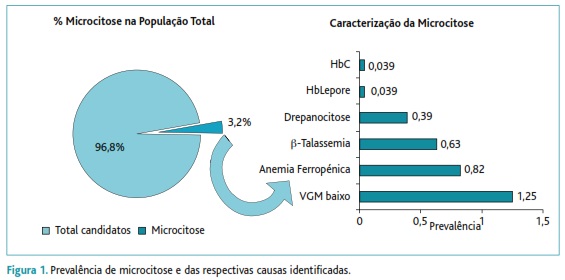
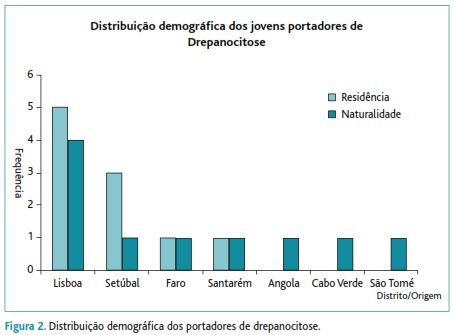
Grupo I: constituído por dez jovens (oito do sexo masculino) portadores de uma heterozigotia para HbS (valor médio de 32,3%±3,46%), maioritariamente nascidos no distrito de Lisboa (40%), seguindo-se os nascidos nas ex-colónias (Angola, Cabo-Verde e São Tomé e Príncipe) e habitantes no distrito de Setúbal (30%) - Figura 2. Comparativamente aos candidatos sem microcitose, este grupo apresentava valores mais baixos de hemoglobina (13,9±2,5g/dL versus 15,2±1,2g/dL), de VGM (73,4±7,41fL versus 90,3±3,7fL) e de hemoglobina globular média (HGM) (23,9±3,23pg versus 30,4±1,2pg). A prevalência de drepanocitose na população total estudada foi de 0,39%.
No Grupo I englobamos também outros dois jovens adultos que apresentavam outras alterações na molécula da hemoglobina. Um é do sexo masculino, residente e nascido em Santarém, portador de uma hemoglobina Lepore (HbA=84,7%, HbA2=2,6%, HbF=2,3% e HbLepore=10,4%) e o outro, do sexo feminino, residente em Lisboa e nascido no Brasil, portador de HbC, (HbC=32,6%, HbA=66,4% e HbA2=1%). A prevalência para a Hb Lepore e HbC foi de 0,039% – Figura 1.
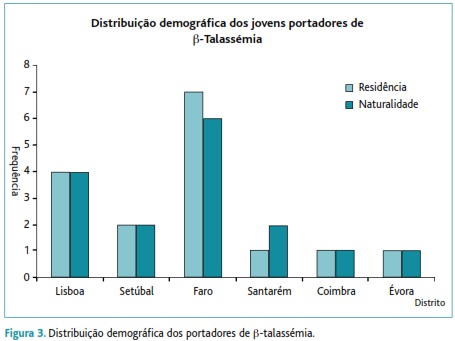
Grupo II: caracterizado por um aumento do doseamento da HbA2 (valor médio de 4,77%±0,63%), ferro e ferritina normais, glóbulos vermelhos (GV) de 6,16±0,51x106/mm3 (versus 5,01±0,37x106/mm3), Hb de 12,7±1,11g/dL (versus 15,2±1,2g/dL), VGM de 64,4±4,57fL (versus 90,3±3,7fL) e HGM de 20,7±1,75pg (versus 30,4±1,2pg). Verificou-se presença de anisocitose, avaliada em lâmina e no hemograma pelo Red Distribuition Widht (RDW=16,5±1,27% versus 12,8±0,9%), de dianócitos ou células em alvo em 56,2% dos jovens e de pontuado basófilo em 68,8%. A prevalência deste grupo associado à β-talassémia foi de 0,63% – Figura 1.
Este grupo era constituído por dezasseis jovens (treze do sexo masculino) sendo 37,5% nascidos e a residir no distrito de Faro, seguindo-se o distrito de Lisboa (25%) e o de Setúbal (12,5%) – Figura 3.
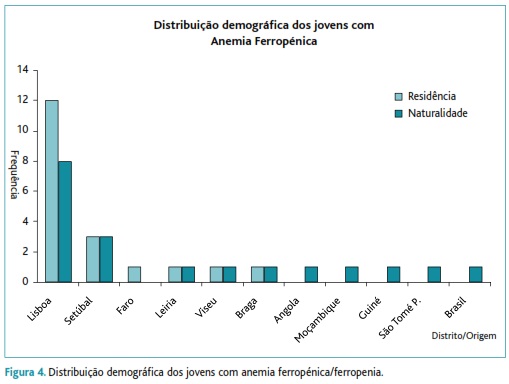
Grupo III: com um total de 21 jovens, dos quais 15 do sexo feminino, maioritariamente habitantes no distrito de Lisboa (57%), tendo 38% nascido neste mesmo distrito e 28% fora de Portugal (Angola, Moçambique, Guiné, São Tomé e Príncipe e Brasil) – Figura 4. Caracteriza-se este grupo por valores mais baixos de ferritina <15 ng/mL (valor médio 6,31±3,11ng/mL versus 93,7±43,9ng/mL), de Hb (11,5±1,96g/dL, versus 15,2±1,2g/dL), de VGM (72,6± 6,39fL versus 90,3±3,7fL) e de HGM (23,7±2,76pg versus 30,4±1,2pg). A anisocitose está presente em 80,9% (RDW=16,3±2,36% versus 12,8±0,9%), seguindo-se a poiquilocitose (52,4%) e eliptocitose (19%). Todos os jovens com ferritina <15ng/mL foram integrados no grupo III, referente aos candidatos com ferropenia. Da análise do valor de hemoglobina podemos constatar que 13 indivíduos (9 do sexo feminino) apresentam valores de hemoglobina <12 mg/dL, portanto com anemia, e 8 (6 do sexo feminino) apenas têm depleção das reservas de ferro. A prevalência deste grupo com anemia ferropénica/ferropenia foi de 0,82% - Figura 1.
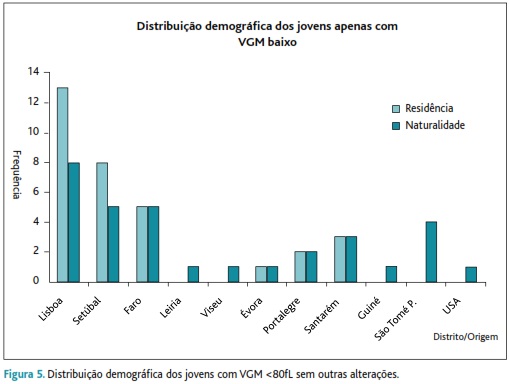
Grupo IV: com 32 jovens que, apesar de terem valores baixos de VGM (76,8±2,27 versus 90,3±3,7fL), não apresentam anemia (Hb=14,3±1,49g/dL) nem alterações na electroforese de Hb. A HGM é variável, encontrando-se a maior parte com valores abaixo da normalidade (25,7±1,34pg versus 30,4±1,2pg). O valor de glóbulos vermelhos está também aumentado (5,58±0,44x106/mm3 versus 5,01±0,37x106/mm3). O ferro e a ferritina estão normais. A conjugação de todos estes parâmetros faz-nos inferir o diagnóstico para uma α-talassémia. Este grupo vive maioritariamente no distrito de Lisboa (43,8%) seguindo-se o de Setúbal (25%) e de Faro (15,6%) – Figura 5. A prevalência desta alteração foi de 1,25% – Figura 1.
DISCUSSÃO DE RESULTADOS
Todos os jovens candidatos a ingressarem no Exército Português que apresentavam microcitose eram assintomáticos e tinham previamente passado nas provas físicas exigidas para o efeito. A caracterização destas microcitoses foi feita com base no hemograma (contagem de glóbulos vermelhos, determinação da hemoglobina e análise dos índices hematológicos – VGM<80fL), no doseamento do ferro e ferritina e na execução da electroforese capilar para estudo da hemoglobina. Os resultados obtidos permitiram separar os candidatos em quatro grupos de patologias distintas: Grupo I, portadores heterozigóticos de drepanocitose e portadores de outras alterações da molécula da hemoglobina, como seja a HbC e a Hb Lepore; Grupo II, portadores de β-talassémia; Grupo III, com anemia ferropénica; e Grupo IV, suspeitos de α-talassémia.
A caracterização e diagnóstico das hemoglobinopatias e talassémias é feita por análise do ADN através de técnicas de biologia molecular. No entanto, o estudo efectuado pode fornecer um diagnóstico presuntivo e parece ter interesse numa abordagem de rastreio numa população jovem em idade reprodutiva com o intuito de fornecer informação que conduza a um aconselhamento e/ou orientação genética.
A electroforese capilar é uma metodologia recente capaz de detectar e separar variantes estruturais de hemoglobina, fazendo o seu doseamento com igual ou melhor precisão que o método convencionado como referência, o HPLC- cromatografia líquida de alta pressão.15,16,17 Alterações estruturais na cadeia da globina capazes de modificar a carga eléctrica total da molécula da hemoglobina são detectadas através da migração na electroforese. Desta forma caracterizámos o grupo I e II. Cotton et al18 descrevem a electroforese capilar como um método de rotina com excelente precisão para a determinação da HbA2. O aumento da HbA2 é o parâmetro mais importante para identificar os portadores de β-talassémia, sendo referido que, quando determinada por electroforese capilar, permite fazer o diagnóstico.18 No entanto, existem casos em que esta não se encontra aumentada e os resultados devem ser interpretados conjuntamente com os parâmetros hematológicos e bioquímicos.19,20 Estão descritos valores que rondam a normalidade de HbA2 em portadores de β-talassémia em situações de anemia ferropénica.21 Assim, todos os jovens caracterizados no grupo III (anemia ferropénica), deveriam corrigir a sua anemia com ferro e fazer novamente uma electroforese de hemoglobina, caso mantenham a microcitose. No entanto, os valores de VGM na talassémia são referenciados como valores mais baixos (VGM = 64,4±4,57 fL) relativamente aos da anemia ferropénica (VGM=72,6±6,39fL) e a contagem total de glóbulos vermelhos está aumentada (6,16±0,51x106/mm3 versus 4,86±0,64x106/mm3) como constatamos também neste estudo. O valor da anisocitose foi muito semelhante nas duas situações. Já a observação do esfregaço de sangue permitiu constatar na β-talassémia a presença de pontuado basófilo, estando este descrito como sendo mais frequente em indivíduos de origem mediterrânea ao invés dos africanos, sendo esta atribuída a uma deficiência da enzima pirimidino 5`nucleotidase (responsável pela degradação do ácido ribonucleico).22,23 Também a presença de dianócitos ou células em alvo são característicos da β-talassémia, consequência da redução do conteúdo citoplasmático sem alteração da quantidade de membrana do glóbulo vermelho. Na anemia ferropénica foi mais constatada a presença de glóbulos vermelhos com formas anormais, destacando-se dentro destas a eliptocitose. Este grupo III é constituído por jovens que maioritariamente apresentam anemia mas existem outros que apresentam só depleção das reservas de ferro.14
A presença de HbS na electroforese de hemoglobina caracterizou os portadores de drepanocitose que, curiosamente e ao contrário do esperado, apresentavam também microcitose. Barbara Bain refere que a existência de microcitose em portadores de drepanocitose advém da existência de outra hemoglobinopatia para além da caracterizada na HbS.23 Pelo facto de a nossa condição de estudo ser um VGM<80fL, não podemos avaliar a prevalência desta patologia na ausência de microcitose nem diagnosticar a causa subjacente, para além do doseamento do ferro e ferritina que caracteriza a existência simultânea de uma anemia ferropénica e a análise da electroforese de hemoglobina que nos aponta para a presença de uma talassémia simultaneamente. A presença de anemia ferropénica foi constatada apenas num jovem também portador de drepanocitose, que apresentava valores de ferritina muito baixos (2,5ng/mL). Noutros dois candidatos, o doseamento da HbA2 estava acima de 3% e abaixo de 4%, o que faz suspeitar da presença simultânea de β–talassémia e drepanocitose. Todos os outros jovens tinham valores normais de ferro e ferritina, bem como doseamentos de HbA2. Estes últimos fazem-nos excluir à priori a existência simultânea de outra hemoglobinopatia com possibilidade de ser detectada na electroforese, fazendo suspeitar por exclusão de partes de uma α-talassémia.
A α-talassémia é uma patologia genética conhecida, mais frequente do que é constatada, pelo que muitos autores a consideram uma doença subdiagnosticada.1,11 A sua presença é suspeita num quadro de microcitose com ou sem anemia, reservas de ferro e electroforese de hemoglobina normais.24 O grau de anemia pode variar e está dependente da alteração genotípica.11 De facto, o grupo de candidatos que corresponde a estes critérios é o que apresenta maior frequência, grupo IV, o que de algum modo nos alerta para a necessidade de caracterizar melhor esta população com vista a fornecer um conhecimento informado.
A Hb Lepore é facilmente identificada na electroforese de hemoglobina capilar pela sua migração, ao contrário da electroforese de hemoglobina convencional e do HPLC17 (nesta a eluição desta hemoglobina é concomitante com a HbA2). Num estudo português é referida como uma hemoglobinopatia comum na população caucasiana habitante no centro do pais.25
A HbC foi também identificada pela electroforese de hemoglobina capilar mas, ao contrário das outras hemoglobinopatias, esta migra numa zona muito próxima da HbA2 dificultando a precisão do seu doseamento. O baixo valor de HbA2 neste caso pode dever-se a uma limitação da metodologia usada como é descrito por Higgins.17
Os portadores silenciosos de α-talassémia, os portadores de traço α e β talassémia, assim como os portadores heterozigóticos de hemoglobinopatias (HbS, HbC, Hb Lepore) são assintomáticos, não requerem tratamento e têm uma esperança média de vida idêntica à da população em geral26 (excepção para os portadores heterozigóticos de HbS, quando submetidos a hipoxia). Reveste-se de grande importância o facto de serem patologias genéticas transmissíveis à descendência com um grau de risco dependente dos progenitores. Assim, se só um dos progenitores for portador de uma talassémia ou de uma hemoglobinopatia, a probabilidade de transmitir a anomalia à descendência é de 25%, aumentando o risco para 50% se ambos forem portadores. Neste caso a probabilidade de nascerem filhos homozigóticos, ou seja, com manifestação de doença grave por talassémia e/ou hemoglobinopatia é também de 50%. É de todo imperativo a identificação dos portadores, assim como de casais em risco, para orientação e aconselhamento genético, evitando desta forma graves problemas sociais e económicos.
A prevalência destas patologias é variável de distrito para distrito, tornando-se premente a existência de rastreios que permitam uma maior vigilância sobretudo nos locais de maior frequência, como sejam Lisboa, Setúbal e Faro. O estudo não permite avaliar a prevalência em Portugal uma vez que só parte do território é rastreada no GCSL. No entanto a existência dos locais descritos na literatura7,8 como tendo maior prevalência são neste estudo também constatados. Por outro lado os números encontrados não nos poderão deixar indiferentes, uma vez que não são patologias assim tão raras.
Concluímos que a avaliação da microcitose nesta população jovem adulta assintomática, feita a partir de uma metodologia de rotina laboratorial simples e pouco dispendiosa, foi suficientemente robusta e capaz de identificar anomalias genéticas e carenciais, contribuindo para um melhor controlo epidemiológico e orientando no sentido de se instituírem medidas preventivas e promotoras de saúde.
AGRADECIMENTOS
Ao Gabinete de Classificação e Selecção de Lisboa.
Ao Sargento-Chefe Carlos Rodrigues e ao 1.o Sargento Luís Lemos agradecemos a colaboração técnica na execução deste trabalho.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Van Vranken M. Evaluation of microcytosis. Am Fam Physician 2010 Nov 1; 82 (9): 1117-22. [ Links ]
2. Moreno Chulilla JA, Romero Colás MS, Gutiérrez Martín M. Classification of anemia for gastroenterologists. World J Gastroenterol 2009 Oct 7; 15 (37): 4627-37. [ Links ]
3. Eicher-Miller HA, Mason AC, Weaver CM, McCabe GP, Boushey CJ. Food insecurity is associated with iron deficiency anaemia in US adolescents. Am J Clin Nutr 2009 Nov; 90 (5): 1358-71. [ Links ]
4. Huang X, Qu X, Yan W, Huang Y, Cai M, Hu B, et al. Iron deficiency anaemia can be improved after eradication of Helicabacter pylori. Postgrad Med J 2010 May; 86 (1015): 272-8. [ Links ]
5. Shams S, Asheri H, Kianmehr A, Ziaee V, Koochakzadeh L, Monajemzadeh M, et al. The prevalence of iron deficiency anaemia in female medical students in Tehran. Singapore Med J 2010 Feb; 51 (2): 116-9. [ Links ]
6. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ 2001; 79 (8): 704-12. [ Links ]
7. Martins MC, Olim G, Melo J, Magalhães HA, Rodrigues MO. Hereditary anaemias in Portugal: epidemiology, public health significance, and control. J Med Genet 1993 Mar; 30 (3): 235-9. [ Links ]
8. Inez F, Sequeira M, Santos P, Santos R, Nunes E et al. Contribuição do rastreio de portadores para a prevenção da β-Talassémia e da drepanocitose na população portuguesa: um estudo multicêntrico. Arq INSA 1993; 19: 27-31. [ Links ]
9. Clarke MG, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem 2000 Aug; 46 (8 Pt 2): 1284-90. [ Links ]
10. Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis 2010 May 21; 5: 11. [ Links ]
11. Harteveld CL, Higgs DR. α-Thalassaemia. Orphanet J Rare Dis 2010 May 28, 5: 13.
12. Chaibunruang A, Srivorakun H, Fucharoen S, Fucharoen G, Sae-ung N, Sanchaisuriya K. Interactions of hemoglobin Lepore (deltabeta hybrid hemoglobin) with various hemoglobinopathies: a molecular and haematological characteristics and differential diagnosis. Blood Cells Mol Dis 2010 Mar 15; 44 (3): 140-5. [ Links ]
13. Srinoun K, Svasti S, Chumworathayee W, Vadolas J, Vattanaviboon P, Fucharoen S, et al. Imbalanced globin chain synthesis determines erythroid cell pathology in thalassemic mice. Haematologica 2009 Sep; 94 (9): 1211-9. [ Links ]
14. WHO UNICEF, UNU. Iron deficiency anaemia: assessment, prevention and control. A guide for programme managers. Geneva: World Health Organization; 2001. Disponível em:http://whqlibdoc.who.int/hq/2001/WHO_NHD_01.3.pdf [acedido em 23/11/2011]. [ Links ]
15. Winichagoon P, Svasti S, Munkongdee T, Chaiya W, Boonmongkol P, Chantrakul N, et al. Rapid diagnosis of thalassemias and other hemo-globinopathies by capillary electrophoresis system. Transl Res 2008 Oct; 152 (4): 178-84. [ Links ]
16. Mais DD, Gulbranson RD, Keren DF. The range of hemoglobin A2 in hemoglobin E heterozygotes as determined by capillary electrophoresis. Am J Clin Pathol 2009 Jul; 132 (1): 34-8. [ Links ]
17. Higgins TN, Khajuria A, Mack M. Quantification of HbA2 in patients with and without β-Thalassemia and in the presence of HbS, HbC, HbE, and HbD Punjab hemoglobin variants: comparison of two systems. Am J Clin Pathol 2009 Mar; 131 (3): 357-62. [ Links ]
18. Cotton F, Lin C, Fontaine B, Gulbis B, Janssens J, Vertongen F. Evaluation of a capillary electrophoresis method for routine determination of hemoglobins A2 and F. Clin Chem 1999 Feb; 45 (2): 237-43. [ Links ]
19. Giambona A, Passarello C, Renda D, Maggio A. The significance of the hemoglobin A(2) value in screening for hemoglobinopathies. Clin Biochem 2009 Dec; 42 (18): 1786-96. [ Links ]
20. Mosca A, Paleari R, Ivaldi G, Galanello R, Giordano PC. The role of haemoglobin A(2) testing in the diagnosis of thalassaemias and related haemoglobinopathies. J Clin Pathol 2009 Jan; 62 (1): 13-7. [ Links ]
21. El-Agouza I, Abu Shahla A, Sirdah M. The effect of iron deficiency anaemia on the levels of haemoglobin subtypes: possible consequences for clinical diagnosis. Clin Lab Haemotol 2002 Oct; 24 (5): 285-9. [ Links ]
22. Vives Corrons JL, Miguel-García A, Pujades MA, Miguel-Sosa A, Cambiazzo S, Linares M, et al. Increased susceptibility of microcytic red blood cells to in vitro oxidative stress. Eur J Haematol 1995; 55 (5): 327-31. [ Links ]
23. Bain BJ. Haemoglobinopathy Diagnosis. London: Blackwell Science; 2001. p. 118. [ Links ]
24. Bryant BJ, Hopkins JA, Arceo SM, Leitman SE. Evaluation of low red blood cel mean corpuscular volume in an apheresis donor population. Transfusion 2009 Sep; 49 (9): 1971-6. [ Links ]
25. Ribeiro ML, Cunha E, Gonçalves P, Martín Nuñez G, Fernandez Galan MA, Tamagnini GP, et al. Hb Lepore-Baltimore (delta 68Leu-beta 84Thr) and Hb Lepore-Washington-Boston (delta 87Gln-beta IVS-II-8) in central Portugal and Spanish Alta Extremadura. Hum Genet 1997 May; 99 (5): 669-73. [ Links ]
26. Muncie HL Jr, Campbell J. Alpha and beta thalassemia. Am Fam Physician 2009 Aug; 80 (4): 339-44. [ Links ]
CONFLITOS DE INTERESSE
Os autores declaram não possuir qualquer tipo de conflitos de interesse.
Manuela Rebordão
Rua Professor Manuel Cavaleiro Ferreira, 4C - 1.o A
1600-642 Lisboa
Recebido em 07/03/2011
Aceite para publicação em 27/10/2011
