








Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkCorrosão e Protecção de Materiais
On-line version ISSN 2182-6587
Corros. Prot. Mater. vol.32 no.2 Lisboa June 2013
ARTIGO
Uma introdução à técnica svet
An introduction to the svet
A. C. Bastos(1)*, S. A. S. Dias(2), T. C. Diamantino(2) e M. y. S. Ferreira(1)
(1) Universidade de Aveiro, Departamento de Engenharia de Materiais e Cerâmica, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
(2) LNEG – Laboratório Nacional de Energia e Geologia, Laboratório de Materiais e Revestimentos (LMR), Estrada do Paço do Lumiar, 22, Ed. E, 1649-038 Lisboa, Portugal
(*) A quem a correspondência deve ser dirigida: acbastos@ua.pt
RESUMO
Este artigo apresenta uma introdução à Técnica do Eléctrodo Vibrante de Varrimento (SVET). Começa por uma breve resenha histórica, seguida da descrição do princípio de funcionamento, exemplos da aplicação a casos de corrosão, alguns cálculos possíveis, limitações, instrumentação, detalhes experimentais e exemplos da sua associação a outras técnicas electroquímicas.
Palavras-chave:: SVET, SRET
ABSTRACT
This paper presents an introduction to the SVET (Scanning Vibrating Electrode Technique). It starts with a brief historical account, followed by the operating principle, examples of application to corrosion, possible calculations, limitations, instrumentation, technical details and examples of its association with other electrochemical techniques.
Keywords: SVET, SRET
1. INTRODUCTION
A técnica SVET (do nome inglês Scanning Vibrating Electrode Technique, que em português pode ser traduzido por Técnica do Eléctrodo Vibrante de Varrimento) foi originalmente desenvolvida por biólogos entre as décadas de 1950 e 1970 com o objectivo de medir as correntes iónicas associadas a processos de diferenciação celular, regeneração de tecidos e electrofisiologia, áreas onde é conhecida como Vibrating Probe [1-8]. A sua introdução na área da corrosão ocorreu na década de 1980 por Hugh Isaacs [9-12]. Até então, o potencial e a corrente em solução eram medidos usando eléctrodos de referência, geralmente com capilares de Luggin-Haber, ideia iniciada no final dos anos 1930s por Thornhill e Evans [13-14] e, principalmente, Agar e Evans [15-16].
O conceito teve continuidade nas décadas seguintes por vários autores, destacando-se as referências [17-23]. A primeira revisão bibliográfica surgiu em 1965 [24] e em 1981 Isaacs faz uma nova revisão [25] tendo então denominado de SRET (Scanning Reference Electrode Technique) o conjunto de técnicas de mapeamento baseadas em eléctrodos de referência não vibrantes. Pode ainda referir-se o livro de Helmut Kaesche [26] onde muitos dos estudos referidos atrás foram analisados em detalhe. Revisões recentes de trabalhos publicados usando SRET e SVET podem ser encontradas em [27, 28]. A sua consulta revela que estas técnicas têm sido utilizadas na análise de casos de corrosão galvânica, corrosão por picada, corrosão sob tensão, corrosão intergranular, corrosão microbiológica, inibidores de corrosão e revestimentos.
2. PRINCÍPIO DE FUNCIONAMENTO
Provavelmente a melhor forma de entender o funcionamento da SRET e da SVET na medição do potencial e da corrente em solução é considerar uma célula com placas paralelas como a que se apresenta na figura 1a) por onde circula uma corrente contínua de 100 µA. A figura 1b) mostra a diferença de potencial medida em vários pontos do circuito, com destaque para a queda óhmica em solução, a qual se reduz apreciavelmente quando a concentração (e consequentemente a conductividade) da solução aumenta. O perfil da queda óhmica foi obtido também com um microeléctrodo de referência que se moveu relativamente a outro mantido fixo - figura 1c). A variação de potencial em solução foi cerca de 120 mV cm-1 para a solução NaCl 5 mM e 1,5 mV cm-1 para a solução cem vezes mais concentrada e com uma conductividade 78 vezes maior. A densidade de corrente em solução, i, pode ser calculada usando a seguinte equação,

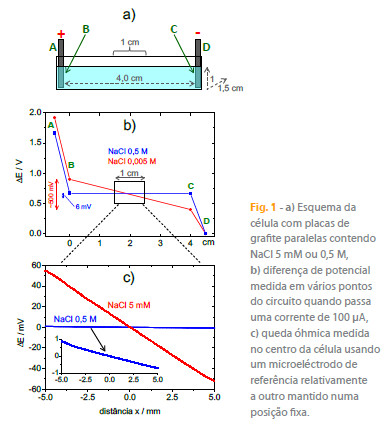
onde k é a conductividade da solução, E é o campo eléctrico na solução e ?V é a diferença de potencial entre duas posições em solução distanciadas por ?r e alinhadas perpendicularmente com as placas de grafite. A figura 2a) apresenta a densidade de corrente obtida para as duas soluções, usando ?r = 20 µm. Embora com bastante ruído, de forma particulamente evidente para a solução de 0,5 M NaCl, o valor médio é semelhante e pode ser comparado com o valor teórico, que é de 100 µA / 1,5 cm2 = 66,(6) µA cm-2 para a geometria de célula usada.
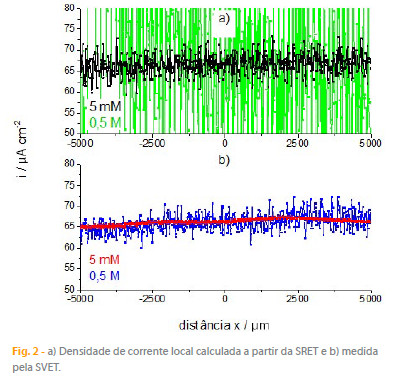
O que se acaba de descrever é um exemplo de medida SRET. Uma variante desta técnica emprega dois eléctrodos de referência (por vezes eléctrodos de ouro ou de platina com negro de platina) com posições fixas e ?r em geral entre 10 e 100 µm [22, 29]. A diferença de potencial entre os dois eléctrodos é dada imediatamente e a densidade de corrente i é obtida pela multiplicação por k. Outra variante da SRET consiste na modificação do sistema para o estudo de amostras cilíndricas [30, 31].
Normalmente, a densidade de corrente dada pela SRET apresenta bastante ruído, um problema que se agrava com o aumento da conductividade do meio usado. Pode-se reduzir significativamente o ruído e aumentar a sensibilidade da técnica fazendo o eléctrodo vibrar. Com a vibração o sinal medido passa a apresentar uma forma sinusoidal susceptível de ser amplificada e filtrada num amplificador lock-in. Esta é a base da técnica SVET e a principal diferença em relação à SRET. O fundamento continua a ser uma medida de potencial, ?V, entre os extremos da vibração do eléctrodo, ?r, usando-se a equação (1) para obter a densidade de corrente. A figura 2b) apresenta a corrente medida pela SVET nas condições experimentais anteriores verificandose um valor de corrente muito próximo do teórico mas com bastante menos ruído quando comparado com os resultados SRET.
Ambas as técnicas dão a conhecer as correntes iónicas a partir de medidas do potencial em solução. A SVET tem como principais vantagens a maior sensibilidade e menor ruído. Por outro lado, como o eléctrodo da SRET não vibra e como pode ter uma extremidade bastante mais pequena (entre 0,1 e 1 µm, enquanto o eléctrodo da SVET raramente é inferior a 10 µm), pode aproximar-se mais das fontes de corrente permitindo melhor resolução espacial.
3. EXEMPLOS DE UTILIZAÇÃO EM CORROSÃO
Na figura 1, ânodo e cátodo encontram-se frente a frente. Em corrosão dispõem-se lado a lado, com as correntes em solução saindo e entrando na mesma superfície. Um exemplo é o par galvânico zinco-ferro que se apresenta nas figura 3 a) e 3 b). Quando ligados electricamente num meio electrolítico condutor, à temperatura ambiente, o zinco oxida-se protegendo o ferro que funciona como cátodo e onde o oxigénio dissolvido em solução se reduz com a produção de OH-. O movimento de electrões na fase metálica do zinco para o ferro é acompanhado na fase aquosa por um fluxo de aniões para o ânodo (zinco) e de catiões para o cátodo (ferro). As figuras 3 c) e d) mostram, respectivamente, mapas de potencial em solução (medido com um microeléctrodo de referência) e mapas de correntes iónicas (medidas com a SVET) obtidos nos planos xy e xz em relação à superfície da amostra. A SVET detecta o fluxo ascendente dos catiões resultantes do processo anódico como corrente positiva (a vermelho nos mapas) e o fluxo ascendente de aniões (OH-) do processo catódico como corrente negativa (a azul nos mapas). Na realidade, todas as espécies carregadas transportam a corrente e são responsáveis pelo sinal medido pela SVET. Junto aos eléctrodos, dentro da chamada camada de difusão, é importante a contribuição do Zn2+ e do OH-.
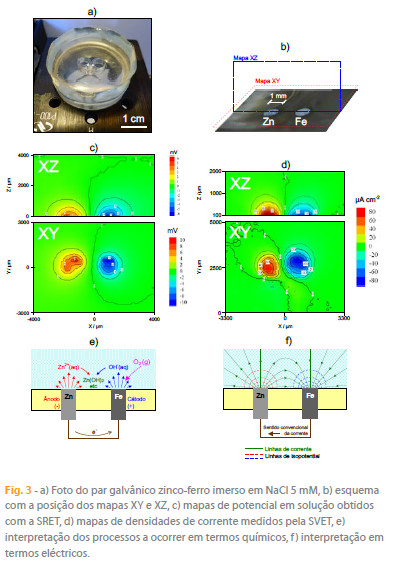
A fracção de corrente transportada pelo Na+ e pelo Cl- (iões que constituem a solução de ensaio) aumenta com a distância à superfície até que no seio da solução a corrente é transportada essencialmente por estes iões. As figura 3 e) e 3 f) mostram ainda interpretações química e eléctrica dos processos a ocorrer. Os esquemas não estão à escala e devem ser entendidos apenas a título meramente ilustrativo.
Na célula usada, zinco e ferro foram dispostos de forma a ter-se os processos anódico e catódico separados e bem localizados a fim de se entender facilmente os resultados fornecidos pelas duas técnicas. Uma combinação mais comum destes dois metais é o revestimento de aço por camadas de zinco. A figura 4 mostra um aço electrozincado após 24 horas de imersão em NaCl 0,05 M e várias formas de apresentar o mesmo resultado obtido pela SVET. A corrosão do zinco ocorreu de forma localizada atingindo a base de aço em vários pontos da superfície. O equipamento SVET utilizado permite a medição do campo eléctrico em duas direcções (x e z) o que permite a apresentação da corrente na forma de vectores 2D. Em corrosão, no entanto, é costume utilizar apenas a componente da corrente perpendicular à superfície da amostra pois os mapas resultantes dão uma boa ideia da distribuição espacial dos processos anódico e catódico.
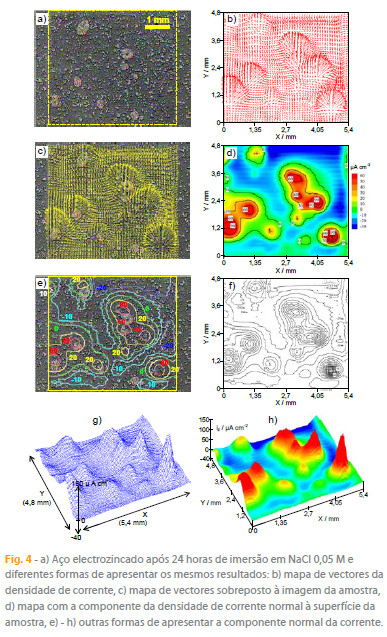
Amostras homogéneas de um mesmo metal puro ou liga também podem ser estudadas pela SVET desde que as regiões anódicas e catódicas sejam de dimensões passíveis de ser resolvidas pela técnica. Se a corrosão não for muito rápida, é possível seguir a evolução espacial e temporal dos processos durante a degradação da amostra, como se exemplifica na figura 5, com vários instantes da corrosão de uma amostra de aço macio durante as primeiras 24 horas de imersão em NaCl 0,05 M.
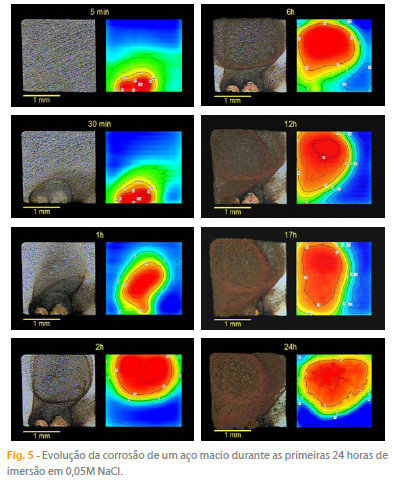
A SVET pode ser vista como uma técnica de visualização da corrosão e poder-se-ia argumentar que o mesmo é conseguido simplesmente com uma câmara que registe a evolução do processo ao longo do tempo. A figura 5 mostra, no entanto, de forma clara, algumas das vantagens da SVET como forma de visualização complementar à imagem óptica. A SVET detecta a actividade antes dela se revelar a olho nú ou ao microscópio. Note-se como aos 5 minutos de imersão a imagem óptica apresenta a superfície ainda intacta enquanto a SVET já detecta os processos que estão a ocorrer, sendo o resultado destes apenas visível mais tarde (compare-se o mapa SVET aos 5 minutos com a superfície da amostra aos 30 minutos). Por outro lado, as imagens ópticas mostram o processo corrosivo acumulado até ao momento da sua aquisição sem distinguir as partes da superfície que estão activas nesse instante. A SVET, por seu lado, mostra a actividade instantânea, ou seja, aquela que de facto ocorre durante a obtenção do mapa (5 a 30 minutos são tempos típicos, dependendo no número de pontos e do tempo de integração em cada ponto), distinguindo as zonas anódicas e catódicas e dando uma medida semi-quantitativa sobre as respectivas actividades.
4. ALGUNS CÁLCULOS
Como o demonstram os resultados anteriores, a SVET é uma técnica bastante útil para a visualização dos processos corrosivos, mas por vezes há o interesse em explorar um pouco mais e tentar usar os resultados para obter informação quantitativa e, inclusivamente, estimar a corrente de corrosão. Apresenta-se nesta secção alguns dos cálculos possíveis e as limitações inerentes aos resultados da SVET, utilizando a figura 6 como exemplo.
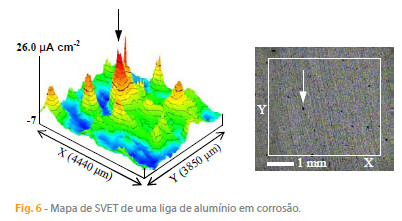
4.1. Corrente que passa num ponto medido pela SVET
A densidade de corrente no ponto indicado na figura 6 é de 26 mA cm-2. Significa que o campo eléctrico sentido pela SVET nesse ponto é igual ao campo eléctrico gerado por uma corrente de 26 mA que passa numa área de 1 cm2 num meio com a mesma conductividade. Para determinar a corrente absoluta que passa nesse ponto é necessário conhecer a área correspondente ao ponto de medida, que pode ser obtida dividindo a área do mapa pelo número de pontos. Neste exemplo, a área do mapa é 0,3850 cm x 0,4440 cm = 0,1710 cm2 e o número de pontos é 1600 (40 x 40), pelo que área corespondente a cada ponto é 1,07 x 10-4 cm2. A corrente absoluta que passa naquele ponto é portanto (26 mA cm-2) x (1,07 x 10-4 cm2) = 0,00278 mA (2,78 nA). Um número maior de pontos aumenta a resolução do mapa. A corrente total, correspondente à área total do mapa, obviamente, permanece a mesma.
4.2. Correlação entre a corrente medida em solução com a corrente na fonte que lhe dá origem
A SVET mede as correntes iónicas em solução que atravessam um plano que se encontra tipicamente a 100-200 mm acima da superfíce metálica (plano de medição). Em geral não é essa corrente que se pretende conhecer mas sim a corrente que atravessa a interface metal-solução. Não é fácil correlaccionar as duas quantidades. De facto, a correlação só é simples para uma fonte de corrente pontual, ou seja, um ponto diminuto que debita a corrente para um contra-eléctrodo distante. Se a fonte pontual de corrente estiver embutida numa superfície plana não condutora, a corrente I debitada pela fonte que origina a densidade de corrente medida pela SVET, iSVET, pode ser calculada por,

onde r é a distância da sonda à superfície. Se se admitir que o pico de densidade de corrente considerado na figura 6 é criado apenas por uma única picada, rodeada por uma área catódica, sabendo que o mapa foi obtido a 200 µm da superfície, pode-se tentar usar a equação (2): I = (2) (p) (200 x 10-4 cm)2 (26 mA cm-2) = 65 nA. Esta é a corrente absoluta, gerada naquele ponto da superfície metálica, que dá origem à densidade de corrente medida pela SVET naquele ponto do mapa. Este cálculo estaria correcto se só houvesse aquele ponto activo na superfície. No entanto, toda a amostra está activa e a corrente da amostra corresponde à actividade de todos os pontos da superfície. Há pontos com actividade anódica, ou catódica, com intensidades distintas, pontos sem actividade, pontos isolados e pontos agrupados em regiões com a mesma actividade.
4.3. Corrente na superfície da amostra
A determinação da corrente em toda a amostra a fim de obter um valor de corrente de corrosão que possa ser comparado com os valores obtidos por outras técnicas é uma tarefa difícil que requer simulação numérica. Em alternativa, vários autores têm tentado uma abordagem aproximada e falível. O procedimento consiste no somatório das correntes (não densidades de corrente) de todos os pontos positivos (ou todos os pontos negativos) de onde resulta a corrente da amostra. A divisão da corrente total pela área da amostra dá a densidade da corrente da amostra. As correntes positivas e negativas assim obtidas deverão cancelar-se.
5. LIMITAÇÕES
O procedimento descrito em 4.3. raramente conduz a resultados aceitáveis por quatro razões principais:
i) As medidas não são feitas na superfície do metal mas a uma determinada distância acima dela. A técnica não contabiliza a corrente que flui entre ânodos e cátodos abaixo dessa altura, como esquematizado na figura 7 a) .
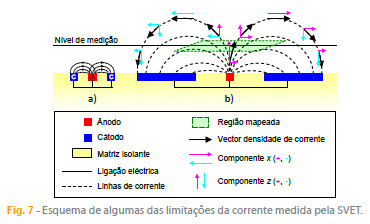
ii) A corrente que ascende e atravessa o plano de medição regressa à superfície atravessando de novo esse plano em sentido contrário. O regresso poderá ocorrer fora da região mapeada e essa corrente não é medida, como se exemplifica na figura 7 b) . É o caso de correntes anódicas localizadas e muito intensas rodeadas por áreas catódicas bem maiores e com correntes pequenas.
iii) A terceira razão está relacionada com a sensibilidade da técnica. O nível de ruído da SVET na gama de soluções típicas de ensaio (0,01 - 0,1 M) é cerca de 1 µA cm-2. Significa que correntes inferiores não são detectadas. As correntes de grandes áreas catódicas apresentam valores dessa ordem de grandeza ou mesmo inferiores.
iv) A última razão costuma ser esquecida: a corrente flui nas três direcções, mas habitualmente só se usa a componente z da densidade de corrente (figura 7 b) , pelo que a corrente medida é quase sempre subestimada e raramente as correntes positivas e negativas se cancelam.
Torna-se evidente que embora a SVET possa ser usada para estimar velocidades de corrosão, os valores estimados têm associado um elevado grau de incerteza. A fiabilidade desses valores necessita de ser confirmada por um segundo método, tipicamente curvas de polarização ou espectroscopia de impedância electroquímica, o que torna a SVET redundante e escusada para esse fim.
Estas limitações permitem concluir que a SVET deve ser vista como uma técnica semi-quantitativa para visualização da corrosão, não um método para determinar velocidades de corrosão.
6. MODELAÇÃO E SIMULAÇÃO
Para ultrapassar as limitações referidas na secção anterior é necessário recorrer a simulação numérica. Esta é necessária mesmo para uma geometria aparentemente simples como um disco. Isaacs reviu e comparou equações para o caso de uma fonte pontual de corrente, um disco de corrente uniforme e um disco de potencial constante [32]. Os resultados das equações para os três casos coincidem quando a distância à superfície é superior a duas vezes o raio do disco. As diferenças para distâncias inferiores podem ser entendidas à luz da distribuição da corrente, primária, secundária ou terciária [33, 34].
A distribuição primária, a mais simples de tratar matematicamente, depende unicamente da geometria da célula, incluindo o tamanho e forma dos eléctrodos, mas normalmente só é aplicável a fontes pontuais de corrente ou quando se analisam regiões em solução afastadas dos eléctrodos. Na maioria dos casos a polarização dos eléctrodos determina a distribuição espacial do potencial e da corrente e tem que ser tida em conta nos cálculos. Nestas condições a distribuição é chamada de secundária. Quando a sonda está bastante perto da superfície, há ainda a considerar os gradientes de concentração das espécies carregadas geradas ou consumidas no eléctrodo que só são devidamente contabilizados quando se considera a distribuição terciária. Excepto para casos muito simples, é necessário recorrer a simulação numérica para relacionar os processos na superfície com os que tem lugar em solução. Existem pacotes comerciais, como por exemplo o COMSOL Multiphysics ou o MAT LAB, que podem ser usados na simulação destes casos, recorrendo ao método dos elementos finitos ou dos elementos fronteira. Cabe ainda referir aqui um conjunto de artigos publicados nos anos 50 do século XX onde se analisou matematicamente a distribuição de corrente e potencial em solução para vários tipos de célula (Wagner, [35]) e casos de corrosão (Waber, [36]).
7. INSTRUMENTAÇÃO E DETALHES EXPERIMENTAIS
Há apenas dois instrumentos SVET disponíveis no mercado, um da empresa Uniscan [37] e outro da empresa Applicable Electronics Inc. [38]. A japonesa Hokuto-Denko chegou a comercializar um modelo na década de 1990. Além destes, vários equipamentos foram desenvolvidos no passado pelos próprios investigadores [3-5, 39-41].
Todos os resultados apresentados neste artigo foram obtidos com o equipamento da Applicable Electronics (AE). A informação que se segue refere-se apenas a esse aparelho porque os autores não conhecem detalhes sobre a operação do equipamento da Uniscan. Embora o princípio de funcionamento seja o mesmo, os detalhes técnicos são certamente diferentes. Talvez as principais diferenças entre os dois sejam a dupla vibração no caso da AE, o tamanho das áreas mapeadas (milímetros para a AE, centímetros para a Uniscan) e a maior facilidade de conjugar o equipamento da AE com outras técnicas e aparelhos.
O equipamento da AE foi desenvolvido para aplicações em biologia e baseia-se no instrumento descrito em [5]. Na figura 8 a) mostra-se o eléctrodo vibrante, geralmente uma haste metálica de platina-irídio (80%/20%) com 1,5 cm de comprimento e 250 µm de diâmetro, afilada na extremidade e revestida por uma camada de 3 µm de parylene-C, um polímero aplicado por deposição química em fase vapor. A extremidade do eléctrodo tem cerca de 3 µm e é o único ponto onde o revestimento foi removido, por intermédio de um arco voltaico. Estes eléctrodos são produzidos pela empresa Microprobes [42] e a principal aplicação é a neuroestimulação. Para poderem ser utilizados como eléctrodos SVET é necessário aumentar a área electricamente activa, o que é conseguido pela formação de um depósito de negro de platina electrodepositado a partir de ácido hexacloroplatínico. A célula electroquímica típica para uma medida SVET encontra-se na figura 8 b) , com a superfície da amostra isolada, excepto a área a mapear, colada num suporte não condutor de 3 cm de diâmetro e fita adesiva em redor do suporte, onde se coloca a solução de ensaio. A medição de potencial ocorre entre o eléctrodo vibrante e um fio de platina com negro de platina que funciona como referência. Um segundo fio de platina platinizado é a ligação à t erra do transistor de efeito de campo de dupla junção ( JFET U401) que se encontra num pré-amplificador e que é o centro da medição SVET.
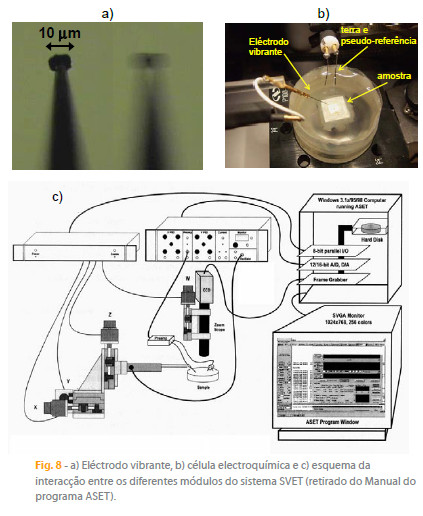
O sistema completo está esquematizado na figura 8 c) e consiste numa câmara de vídeo que permite controlar a posição do eléctrodo e obter imagens da amostra, um sistema de motores que posiciona e movimenta o microeléctrodo com 1 µm de precisão, um pré-amplificador e um amplificador IPA-2 que integra dois amplificadores lock-in, um para cada vibração. O microeléctrodo é colocado na extremidade de um braço de plástico estando a outra extremidade ligada a dois osciladores piezoelectrónicos que são responsáveis pelas vibrações x e z (frequências seleccionáveis entre 40 e 1000 Hz). Todo o processo é controlado pelo software ASET desenvolvido pela empresa Sciencewares [43]. Uma mesa anti-vibração, uma gaiola de Faraday e uma unidade de alimentação ininterrupta com filtragem da corrente de alimentação são opcionais mas recomendadas para o óptimo desempenho.
Como o sistema mede diferenças de potencial e apresenta os resultados na forma de densidades de corrente, é necessária uma calibração prévia para relacionar as duas quantidades. Para tal, a sonda é posicionada a uma determinada distância (geralmente 150 µm) de uma fonte de corrente pontual que debita uma corrente I conhecida (geralmente 60 nA). A densidade de corrente i à distância r

O sistema mede a diferença de potencial e determina o factor de proporcionalidade relactivamente à densidade de corrente i. Este factor de proporcionalidade depende da conductividade da solução, da amplificação do sistema, da frequência e da amplitude da vibração. Enquanto a frequência e a amplitude de vibração não forem alteradas a calibração permanece válida sendo apenas necessário introduzir o valor correcto da conductividade da solução em uso. O sistema da AE faz a medição em duas direcções ortogonais. Além disso, o sistema adquire os sinais em fase e em quadratura a fim de compensar quaisquer desvios à perpendicularidade dos eixos de vibração. O cálculo recorre a uma matriz de calibração, como descrito em [5]. Deve referir-se que além desta rotina de calibração, que é a mais comum, existem outras, descritas nas referências [6] e [45].
8. ALGUMAS QUESTÕES OPERACIONAIS
8.1. Sensibilidade
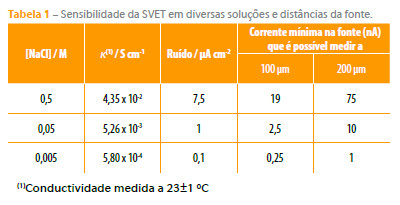
A sensibilidade da SVET, entendida como a menor corrente discriminada acima no nível de ruído, depende directamente da conductividade do meio. Se o que interessa é a corrente mínima na superfície metálica que pode ser detectada pela SVET, então esse valor depende também da distância entre a superfície e o ponto de medida. A Tabela 1 apresenta valores experimentais medidos usando uma fonte pontual de corrente (micropipeta de vidro com uma abertura de 2 µm de diâmetro, cheia com a mesma solução de ensaio, tendo no interior um fio de platina com negro de platina por onde a corrente entra em solução). A SVET consegue medir com fiabilidade correntes em solução que sejam no mínimo duas vezes o nível de ruído. Para os meios de ensaio mais comuns (0,01 - 0,1 M NaCl) a sensibilidade da SVET em solução ronda os 1-2 µA cm-2 a que corresponde uma corrente na fonte de 2 a 10 nA.
8.2. Resolução espacial
Definindo a resolução espacial como a distância mínima entre duas fontes pontuais de igual corrente que conseguem ser distinguidas, pode dizer-se que é difícil de estabelecer um valor concreto pois este depende de vários factores, principalmente da distância à superfície e da conductividade do meio, mas também do tamanho da sonda e da amplitude da vibração. Uma regra é considerar que a SVET só separará o sinal das duas fontes quando se encontrar a uma distância da superfície igual ou inferior a metade da distância entre elas. Um exemplo: para que 2 pontos separados 200 µm sejam discriminados é necessário que a sonda esteja no máximo a 100 µm da superfície. Ainda outro exemplo: Isaacs mostrou [32] que o sinal medido pela SVET acima de um ponto ou de um disco que debitam a mesma corrente absoluta é igual quando a sonda está a uma distância superior a duas vezes o raio do disco. Concretizando: se a sonda se encontrar a 100 µm da super fície, obter-se-á o mesmo mapa quer a fonte de corrente seja um ponto quer seja um disco de 100 µm de diâmetro, desde que a corrente absoluta produzida por ambos seja a mesma. Para os distinguir, a sonda terá de se aproximar da superfície.
8.3. Influência da vibração
Há quatro aspectos a considerar acerca da vibração da SVET:
i) O primeiro refere-se à sua capacidade de agitar a solução homogeneizando localmente a composição química. Esse efeito foi estudado por Lucas e Ferrier [46] que concluiram que os vórtices criados pela vibração do eléctrodo alteram a concentração das espécies químicas na sua vizinhança mas não a densidade de corrente local.
ii) A agitação da solução aumenta o transporte por convecção sendo particularmente importante no transporte do oxigénio dissolvido em solução até à superfície metálica. Este efeito foi estudado por McMurray, Williams e Worsley [47] com um sistema SVET diferente do usado aqui. Na SVET da AE, em medidas feitas a 100 µm, a corrente de redução do oxigénio duplica-se momentaneamente quando a SVET vibra por cima do cátodo [48].
iii) Isaacs estudou a relação entre a amplitude da vibração e a distância média da sonda à superfície e concluiu que quando a sonda se aproxima uma distância inferior a 4 vezes a amplitude da vibração, o sinal medido é sobreestimado [49]. Para a vibração típica da SVET este problema só existe quando a sonda está a menos de 50 µm da superfície, o que raramente se verifica na prática.
iv) Um último aspecto a referir é o risco do não alinhamento do eixo da vibração com a normal à fonte de corrente. Este aspecto já foi estudado [12, 50] e os autores consideraram que desvios até 30º não são significativos. Como referido na secção 7., a calibração do sistema da AE mede o sinal em fase e em quadratura precisamente para compensar desvios à ortogonalidade. Naturalmente, não compensa situações em que a amostra esteja inadvertidamente inclinada ou apresente relevo significativo.
8.4. Análise de subtratos metálicos com revestimentos orgânicos
A aplicação desta técnica na caracterização da corrosão em sistemas de pintura tem merecido grande interesse mas o seu uso é bastante limitado. A SVET só consegue medir a corrente que flui através de defeitos ou descontinuidades no revestimento orgânico. Além disso, se o revestimento for espesso (> 500 µm), a sonda estará bastante afastada da fonte de corrente (superfície metálica) e é possível que não detecte qualquer sinal. Pela mesma razão é raro medir-se algum sinal em empolamentos dos revestimento orgânicos.
9. COMBINAÇÃO DA SVET COM OUTRAS TÉCNICAS
A SVET mede as correntes iónicas em solução mas não dá informação sobre a identidade das espécies envolvidas no processo. Isso poderá ser obtido através de microeléctrodos potenciométricos e amperométricos. Uma vantagem do equipamento SVET da AE é que a adição de apenas um módulo e respectivos microeléctrodos é suficiente para a realização de medidas micropotenciométricas e microamperométricas.
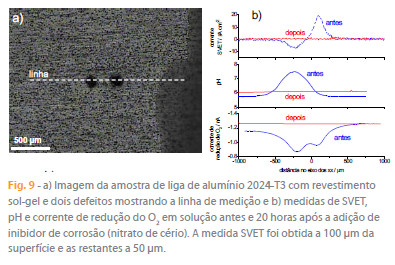
Apresenta-se aqui um exemplo do uso complementar da SVET com a medição potenciométrica de pH e amperométrica do O2 dissolvido em solução. Neste exemplo, amostras da liga de alumínio 2024-T3, liga de aplicação bastante comum em aeronáutica, foram revestidas com um filme produzido pela tecnologia sol-gel. Isolou-se a amostra deixando apenas uma janela de 3 x 3 mm2 exposta à solução e produziram-se dois defeitos no revestimento de modo que o substrato metálico ficasse exposto apenas nesses dois pontos. Deixou-se a amostra corroer em NaCl 0,05 M durante 20 horas após as quais se adicionou um inibidor de corrosão, nitrato de cério. A figura 9 apresenta a corrente em solução, o pH e a corrente de redução de O2 na sonda amperométrica, medidos antes e após a inibição. As correntes iónicas medidas pela SVET antes da inibição mostram um pico positivo sobre um defeito, que se passa a chamar anódico, e um pico negativo sobre o outro defeito, doravante chamado de catódico. O pH da solução aumentou significativamente sobre o defeito catódico (onde se produz OH-) mas não se alterou sobre o defeito anódico. A corrente de redução do O2 foi constante excepto sobre os dois defeitos, onde diminuiu. Esta diminuição deve-se à menor concentração local de O2 uma vez que este é consumido na superfície metálica exposta. Verifica-se que a actividade catódica tem lugar nos dois defeitos, incluindo aquele considerado anódico pela SVET. Existindo O2 em toda a solução e estando a superfície a um potencial onde ele se pode reduzir, a reacção ocorrerá, embora a velocidade de reacção possa ser diferente nos dois defeitos. As medições efectuadas 20 horas após a adição de inibidor mostram valores constantes e iguais aos do seio da solução, o que é interpretado como sendo resultado da inibição, que isolou o metal do meio, impedindo qualquer actividade na superfície da amostra que alterasse a química da solução. O trabalho completo está publicado em [51] e outros exemplos podem ser encontrados nas referências [52-53]. Estes resultados revelam o benefício de combinar as várias técnicas, pois cada uma acresenta uma fracção de informação que reunida dá uma imagem mais completa sobre o sistema em estudo.
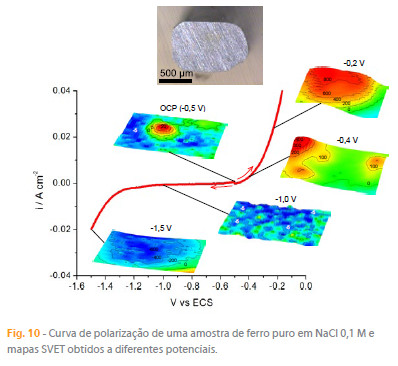
Em todos os exemplos de corrosão apresentados, as amostras corroeram naturalmente, sem aplicação externa de potencial. Contudo, as amostras podem ser polarizadas usando um potenciostato enquanto a SVET mede as correntes resultantes, como mostra a figura 10 para o caso de um eléctrodo de ferro puro com mapas obtidos a vários potenciais. Ao potencial em circuito aberto, potencial de corrosão, o eléctrodo apresenta zonas anódicas e catódicas. Polarizações anódicas ou catódicas promovem, respectivamente, oxidações ou reduções, com a corrente a variar logaritmicamente com a polarização. O mesmo procedimento poderia ser realizado com eléctrodos inertes, como platina por exemplo, em estudos típicos de electroquímica mas, em geral, nesses estudos pretende-se uma distribuição de corrente uniforme (para as quais as medidas de SVET não são interessantes) e adiciona-se electrólito de suporte em abundância para eliminar a componente de migração (precisamente o que é medido pela SVET). Actualmente, quando a informação localizada interessa em estudos de electroquímica, tem-se recorrido à microscopia electroquímica (SECM, Scanning Electrochemical Microscopy) [54].
10. CONCLUSÕES
Pretendeu-se dar a conhecer a técnica SVET e a sua aplicação em estudos de corrosão. Os exemplos apresentados foram criteriosamente seleccionados para uma melhor compreensão do princípio de funcionamento da técnica, as particularidades de que se reveste o seu uso, as vantagens e as limitações. Não se deu ênfase aos detalhes técnicos ou de operação porque muitos deles são inerentes ao instrumento usado e interessam principalmente a quem opera o equipamento. Também não se fez uma revisão bibliográfica dos trabalhos publicados com a SVET. O leitor interessado poderá consultar as referências [27, 28]. Espera-se que estas páginas sejam suficientes para se entender a técnica e as suas especificidades bem como para decidir quando o seu uso é ou não proveitoso. Termina-se reiterando que a SVET deve ser vista como uma técnica de visualização da corrosão, poderosa e versátil, mas vincando que o uso para medidas e cálculos quantitativos deverá ser feito de forma extremamente cuidadosa e esclarecida.
REFERÊNCIAS
[1] O. Bluh, B. I. H. Scott, Rev. Sci. Instrum., 21, 867 (1959). [ Links ]
[2] P. W. Davies, Fed. Proc., 25, 332 (1966). [ Links ]
[3] L. F. Jaffe and R. Nucitelli, J. Cell Biology, 63, 614 (1974). [ Links ]
[4] J. A. Freeman and C. Nicholson, J. Neurophysiol., 38, 369 (1975). [ Links ]
[5] C. Scheffey, Rev. Sci. Instrum., 59, 78 (1988). [ Links ]
[6] B. Reid, R. Nucitelli and M. Zhao, Nature Protocols, 3, 661 (2007). [ Links ]
[7] A. Dorn and M. H. Weisenseel, Protoplasma, 113, 89 (1982). [ Links ]
[8] R. Nucitelli, Experientia, 44, 657 (1988). [ Links ]
[9] Y. Ishikawa and H. S. Isaacs, Boshoku Gijutsu, 33, 147 (1984). [ Links ]
[10] H. S. Isaacs and Y. Ishikawa (Paper 55 - Applications of the vibrating probe to localized current measurements) in Proceedings of NACE Corrosion/85, March, Boston, Massashusetts, USA (1985). [ Links ]
[11] H. S. Isaacs and Y. Ishikawa (Application of the vibrating probe to localized current measurements) in Electrochemical Techniques for Corrosion Engineering (R. Baboian, ed.), National Association of Corrosion Engineers, Houston, Texas, USA, p. 17 (1986). [ Links ]
[12] H. S. Isaacs (Applications of current measurement over corroding metallic surfaces) in Ionic Currents in Development (R. Nuccitelli, ed.), A. R. Liss Inc., New York, p. 37 (1986). [ Links ]
[13] R. S. Tornhill and U. R. Evans, J. Chem. Soc., 614 (1938). [ Links ]
[14] R. S. Tornhill and U. R. Evans, J. Chem. Soc., 2109 (1938). [ Links ]
[15] U. R. Evans, J. Iron and Steel Inst., 141, 219 (1940). [ Links ]
[16] U. R. Evans (The Corrosion and Oxidation of Metals: Scientific Principles and Practical Applications), St. Martins Press, New York, USA, p. 861 (1960). [ Links ]
[17] H. R. Copson, Trans. Electrochem. Soc., 84, 71 (1943). [ Links ]
[18] W. Jaenicke and K. F. Bonhoefer, Z. Phys. Chemie A, 193, 301 (1944). [ Links ]
[19] y. V. Akimov, A. I. Gulobev, Zhur. Fiz. Khim., 20, 303 (1946). [ Links ]
[20] y. Bianchi, La Metallurgia Italiana, 45, 123 (1953). [ Links ]
[21] C. Ilschner-Gensch, Zeitschrift fur Elektrochemie, 64, 275 (1960). [ Links ]
[22] I. L. Rosenfeld and I.S. Danilov, Corros. Sci., 7, 129 (1967). [ Links ]
[23] C. D. S. Tuck, Corros. Sci., 23, 379 (1983). [ Links ]
[24] M. K. Budd, F. F. Booth, Metalloberflache, 19, 197 (1965). [ Links ]
[25] H. Isaacs and B. Vyas (Scanning reference electrode techniques in localized corrosion), in Electrochemical Corrosion Testing, (Mansfeld, Bertocci, eds.), ASTM STP 727, American Society for Testing and Materials, Philadelphia, Pensilvania, USA, p. 3 (1981). [ Links ]
[26] H. Kaesche (Ch. 11 – Galvanic Corrosion Cells) in Corrosion of Metals, Physicochemical Principles and Current Problems, Springer, Berlin, Germany (2003). [ Links ]
[27] R . S. Lillard (Scanning electrode techniques for investigating nearsurface solution current densities) in Analytical Methods in Corrosion Science and Engineering (P. Marcus, F. Mansfeld, eds.), CRC / Taylor & Francis, Boca Raton, Florida, USA, p. 571 (2006). [ Links ]
[28] y. Grundmeier, K.-M. Juttner and M. Stratmann (Novel electrochemical techniques in corrosion research) in Corrosion and Environmental Degradation, Vol. 1, (M. Schutze, ed.), Wiley-VHC, Weinheim, Germany, p. 285 (2000). [ Links ]
[29] K. R. Trethewey, D. A. Sargeant, D. J. Marsh and A. A. Tamimi, Corros. Sci., 35, 127 (1993). [ Links ]
[30] L. J. Gainer and y. R. Wallwork, Corrosion, 35, 61 (1979). [ Links ]
[31] S. R. Allah-Karam, V. Vasantasree and M. y. Hocking, Corros. Sci., 43, 1645 (2001). [ Links ]
[32] H. S. Isaacs, J. Electrochem. Soc., 138, 722 (1991). [ Links ]
[33] J. Newman, K. E. Thomas-Alyea (Electrochemical Systems), 3rd Ed., Wiley, New Jersey, USA, p. 421ss (2004). [ Links ]
[34] N. Ibl, Comprehensive Treatise of Electrochemistry, Vol. 6, (E. Yeager, J. OM. Bockris, B. E. Conway, Eds), Plenum Press (1983). [ Links ]
[35] C. Wagner, J. Electrochem. Soc., 98, 116 (1951). [ Links ]
[36] J. T. Waber, J. Electrochem. Soc, 101, 27 (1954); 102, 344 (1955); 102, 420 (1955); 103, 64 (1956); 103, 138 (1956); 103, 567 (1956).
[37] www.uniscan.com (acesso em 21/3/2013).
[38] www.applicableelectronics.com (acesso em 21/3/2013).
[39] D. Worsley, A. Belghazi and S. M. Powell, Ironmaking Steelmaking, 26, 387 (1999). [ Links ]
[40] J. A. Freeman, P. B. Manis, P. C. Samson and J. P. Wikswo Jr (Microprocessor controlled two- and three-dimensional vibrating probes with video graphics: biological and electro-mechanical applications), in Ionic Currents in Development (R. Nuccitelli, ed.), A. R.Liss Inc., New York, USA, p. 21 (1986). [ Links ]
[41] R . y. Kasper, C. R. Crowe (Comparison of localized ionic currents as measured from 1-D and 3-D vibrating probes with finite-element predictions for an iron-copper galvanic couple) in Galvanic corrosion (H.P. Hack, ed.), ASTM STP 978, American Society for Testing and Materials, Philadelphia, Pensilvania, USA, p. 118 (1988). [ Links ]
[42] www.microprobes.com (acesso em 21/3/2013).
[43] www.sciencewares.com (acesso em 21/3/2013).
[44] C. Scheffey (Electric fields and the vibrating probe, for the uninitiated) in Ionic Currents in Development (R. Nuccitelli, ed.), A. R. Liss Inc., New York, USA, p. xxv (1986). [ Links ]
[45] J. Elvins, J. A. Spittle, J. H. Sullivan and D. A. Worsley, Corros. Sci., 50, 1650 (2008). [ Links ]
[46] J. Ferrier and W. J. Lucas, Biophysical J., 49, 803 (1986). [ Links ]
[47] H. N. McMurray, D. Williams and D. A. Worsley, J. Electrochem. Soc., 150, B567 (2003). [ Links ]
[48] Resultados do autor não publicados.
[49] H. S. Isaacs, Corrosion, 46, 677 (1990). [ Links ]
[50] B. P. Wilson, J. R. Searle, K. Yliniemi, D. A. Worsley and H. N. McMurray, Electrochim. Acta, 66, 52 (2012). [ Links ]
[51] A. C. Bastos, O. V. Karavai, M. L. Zheludkevich, K. A. Yasakau and M. y. S. Ferreira, Electroanalysis, 22, 2009 (2010). [ Links ]
[52] A. C. Bastos, O. V. Karavai, S. V. Lamaka, M. L. Zheludkevich and M. y. S. Ferreira, Corros. Prot. Mater., 28, 42 (2009). [ Links ]
[53] K. Ogle, V. Baudu, L. Garrigues, and X. Philippe, J. Electrochem. Soc., 147, 3654 (2000). [ Links ]
[54] Scanning Electrochemical Microscopy, 2nd Ed (A. J. Bard, M. V. Mirkin, eds.), CRC / Taylor & Francis, Boca Raton, Florida, USA (2012). [ Links ]
Artigo submetido em Março de 2013 e aceite em Junho de 2013